


摘要
代谢组学的气相色谱/质谱(GC/MS)为理解小分子生物学提供了标准化和可靠的平台。自2005年以来,加州大学戴维斯分校西海岸代谢组学中心已将超过156,000个样本和2000项研究的GC/MS代谢组学数据整理到标准化的Binbase数据库中。我们相信,这些样本的观察结果将为生物学家提供有意义的见解,我们的数据处理和网络工具将为其他寻求标准化不同代谢组学研究的人提供见解。我们在这里开发了一个易于使用的查询界面,BinDiscover,使生物学家能够基于这些代谢组学样本直观、快速地生成假设。BinDiscover创建了广泛的物种、器官、疾病和化合物的观察摘要和图形。在整个BinDiscover组件中,我们强调使用本体来基于元数据在这些本体中的接近度来聚合大组样本。这种邻接允许同时探索整个类别,如“啮齿动物”,“消化道”或“氨基酸”。本体论与BinDiscover的本体论分组差异分析特别相关,它与BinDiscover的其他组件一样,可以创建跨化合物和生物元数据的清晰图形和汇总统计。我们在生物领域的三个展示中举例说明了BinDiscover的广泛适用性。
介绍
代谢组学数据库可以用于各种目的。一些数据库将谱库编译成存储库,用户可以下载并合并到他们的识别工作流中。例如北美MassBank (https://massbank.us)[1]或全球天然产品社会分子网络(Global Natural Products Social Molecular Networking, GNPS)[2]。其他例子包括以研究为中心的数据库,存储用户提交的研究的元数据和观察结果,包括代谢组学Workbench[3]、metaboLights和ReDU[3,4,5]数据库。其他的,如人类代谢物数据库(HMDB)[6],可以被粗略地描述为信息编译器,因为它们从一系列来源合成信息。最后(但不是全部)是编译数据库,它聚合了多个较小的数据库。最近的一个例子是椰子(开放天然产品集合)天然产品数据库[7]。代谢组学中最诱人的研究方向之一是协调数据集群岛,以创建临界质量的协同数据,可用于实现对生物学的广泛理解[8]。在metabolomicsWorkbench数据库中,用户可以通过维恩图和代谢物比率数据表查询以代谢物为中心的比较。虽然这样的查询很容易对单个化合物执行,但跨不同研究设计的批量查询的导航界面最好通过需要计算专业知识的应用程序编程接口(api)来执行。HMDB编译来自不同来源的信息。HMDB和来自同一实验室的相关数据库是可靠的,因为这些信息是人工精心整理的。对于HMDB和metabolomicsWorkbench查询,对大量代谢物查询的元分析都会受到影响,因为它是一种回顾性的尝试,旨在协调跨多个生物研究设计的以化合物为中心的信息集。在翻译文本源(如HMDB)或隐式和非结构化的样本/治疗命名方案(如在metabolomicsWorkbench上传期间使用)时,丢失了太多的生物元数据。至少对于复合名称,metabolomicsWorkbench使用了数据库内部命名方案RefMet。然而,由于仪器条件的复杂性和多样性,代谢组学工作台既不知道化合物注释的置信水平,也不知道浓度值。
在个别实验室内,由于在规定的协议下使用特定类型的仪器,数据可能更加协调。在这里,meta-XCMS[9]或Amanida[10]等工具允许生成来自多个研究的结果。然而,这些工具需要特定的输入数据格式,即使在实验室中,当不同的个体处理代谢组学原始数据时,这些数据文件也不是同质的。因此,即使在实验室层面上,以系统的方式收集数据以提供可用于元分析工具的编译结果也不是直截了当的。因此,经典的meta分析是在更高的抽象层次上进行的,例如路径或简化为同义名称集[11],而不是查询大量代谢物表。
我们认识到汇总跨实验室和方法得出的结果的挑战。因此,我们假设,对于研究元数据和数据获取过程而言,协议的标准化是有用的交叉研究比较和查询的关键。与LC-MS /MS相比,GC-MS代谢组学的标准操作程序更加成熟。在加州大学戴维斯分校,自2005年以来,我们使用了一个统一的自动化工作流程来处理代谢组学数据,称为Binbase。我们在这里对2021年冬季之前处理的所有数据进行了快照,以便进行大规模的多研究元分析来调查数据。我们称这个工具为BinDiscover。它是一个网络工具,使用户能够在几分钟内进行元分析,以提取数据趋势并提出假设。我们不是简单地比较两种类型的元数据(例如,具有相同器官的两个不同物种),而是将所有元数据分配到本体中,以便进行更广泛的比较,例如门组织或本体分组差异分析(OGDA)。OGDA查询将广泛的问题转换成一组较小的类别,然后将统计结果输出组合成图。
方法
BinDiscover数据库从GC-Binbase数据库中提取光谱和化合物信息[12,13,14]。GC-Binbase使用桶排序方法,其中来自样品色谱运行的新峰要么与先前注释的分组相匹配,要么被识别为新化合物。这种桶式排序是一种算法,基于脂肪酸甲酯内标(FAME)的保留指数容错为2000个Fiehn RI单位,占大约2秒的绝对保留时间窗口,并匹配在MS反卷积过程中确定的唯一离子。加权点积相似度分数用于使用不同的信号纯度和信号强度阈值将新的实验数据与GC-Binbase中的bin进行匹配[14]。在过去的20年里,所有的复合注释都是手工进行和整理的。GC-Binbase中使用了其他细节,如自动识别“异质干扰”、“峰纯度”、“峰顶离子”、“唯一离子”、“信号/噪声”以及指示数据质量的进一步参数,作为供应商用于质谱反卷积的chroof软件的输出[12,13,14]。在BinDiscover中呈现的光谱是共识光谱,它不断提高分配给Bin的所有单个质谱的光谱质量(具有特定唯一离子和特定保留指数的质谱)。
为了生成BinDiscover数据库,所有分析都是使用Github中可用的自定义python脚本进行的(请参阅“数据可用性”)。我们大量使用来自SciPy的统计例程和来自NetworkX的网络分析框架。开发是在64核、128 gb RAM的Amazon Web Services (AWS)虚拟机上进行全数据转换之前在本地执行的。BinDiscover输出数据库存储在AWS管理的Postgres数据库中。API使用Flask库,前端严重依赖于Plotly/Dash。API和前端通过docker容器化,部署在AWS Elastic Beanstalk上。
结果
Binbase是一个自动数据处理数据库GC-TOF质谱法
在加州大学戴维斯分校西海岸代谢组学中心,初级代谢物研究了18年,使用相同的工作流程进行数据采集和数据处理,使用气相色谱-飞行时间质谱(GC-TOF MS)。目前有5台GC-TOF质谱仪在运行。自2005年以来,标准操作程序已广泛发布,并已锁定并保持不变。用一套脂肪酸甲酯内标对数据进行比对,形成稳定的保留指数。共洗脱质谱通过仪器软件进行去卷积和自动去噪。该软件还提供了一系列关于数据报告质量的元数据,从峰纯度到异构体干扰,绝对和相对离子强度,以及最好地描述其他化合物附近特定代谢物存在的独特离子。所有这些元数据被多级过滤算法用来生成一个综合的已知和未知代谢物数据库,称为Binbase。为了查询交叉研究分析的生物元数据,我们于2021年12月从Binbase下载了所有数据。该数据包括156,174个样品,处理成18,290个bin,即在特定保留时间使用特定定量离子的独特质谱。bin包括773个已鉴定的代谢物,39个已知的化学产物(如GC-TOF MS过程中产生的聚硅氧烷)和15843个未注释为特定化学物质的光谱。在17年的使用过程中,算法伪产物导致多个箱子在数据导出期间合并为单个代谢物值。由于化学三甲基硅基化不完全,一些bin与相同的生物代谢物相关,如以前报道的那样。我们生成了一个工作流程来调查每个Bin的生物关联,称为BinDiscover。一个简化的工作流程如图1所示。GC-TOF MS在Binbase管理图形用户界面(GUI) (BinView)中使用质谱、谱相似性和库谱之间的保留指数差异以及计算保留时间进行化合物鉴定。对于化合物鉴定,FiehnLib文库[12]与MassBank一起使用。us和NIST20光谱[15]。Kovats保留指数值(基于烷烃洗脱顺序)自动归一化为基于脂肪酸甲酯(FAME)洗脱顺序的Fiehn保留指数值。
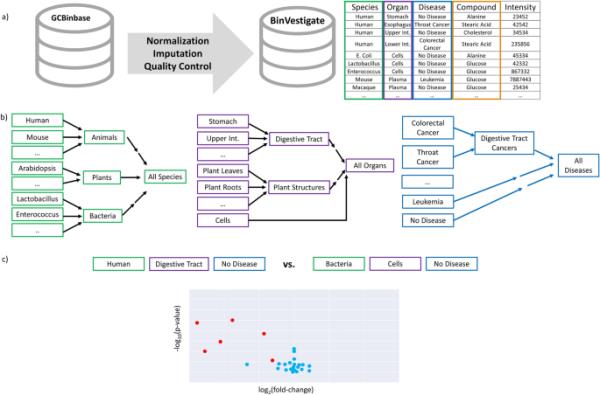
BinDiscover数据库查询的整体工作流程。从2005年到2021年,Binbase在GC-TOF质谱仪上记录了156174份代谢组学样本的观察结果。对相应的生物元数据进行整理,生成的注释表构成了探索性web工具BinDiscover的基础。b: discover关联了跨物种、器官和疾病的代谢物强度。已建立的本体用于为查询排序生物元数据。对于代谢物,我们使用classsyfire本体来启用复合类级查询。c生物元数据与所有样本相关联,可以通过不同的本体级别(如“消化系统”或“细菌”)进行表示和查询。物种、器官和疾病本体用颜色突出显示
争吵和改变我生理上和生理上的我tadata
每个Bin都与所有研究中阳性检测到的生物信息相关联。生物元数据如下所示,将样本元数据映射到已建立的本体。我们使用了三个本体:(1)国家生物技术信息中心(NCBI)的物种分类[16,17],(2)器官和疾病的医学主题标题(MeSH)分类[18],以及化合物的ClassyFire本体[19]。我们总共使用和输入了1696个元数据组合,定义为特定的器官/物种/疾病三元组。在所有样品中,共有55,261,308种观察到的代谢物与Bin相关,以及每个特定Bin的全光谱和定量离子强度。Binbase中的每个样本都与所进行的相应生物学研究的信息相关联。研究包括已发表的和未发表的实验,因为数据是为过去18年的内部学术目的以及外部收费服务项目收集的。将生物元数据输入到SetupX的小版本miniX中[13]。客户输入最少的信息,如物种、器官、简短摘要和样本标签,其中包含研究设计的特定方面的文本。由于没有通用的算法以连贯和机器可读的形式捕获生物设计的所有细节,生物元数据必然保持异构。因此,我们必须整理生物元数据,转换并规范离子强度。
第一步是消除在过去17年中使用4台GC-TOF质谱仪和不同仪器条件所产生的技术差异。在所有研究中,使用了完全相同浓度的FAME内标,使我们有机会使用FAME保留指数标记物作为每个特定样品的仪器性能的替代值。因此,我们将每个样品中的代谢物强度通过FAME离子强度的总和归一化。我们验证了FAME强度在所有样品中的相关性大于0.8,表明它们也反映了GC-MS注射条件的差异。接下来,我们自动识别有问题的样本,并从BinDiscover中排除这些样本。为此,我们删除了具有较差FAME模式的样本,即定义为极高或极低FAME强度值的样本。此外,我们删除了整个生物元数据三元组,如果它们显示超过20%的失败FAME样本(附加文件3:图S1),或者如果特定生物元数据三元组的样本总数少于10个。这种数据争论确保了异常值对任何特定生物类别的平均代谢物强度没有过大的影响。通过这种方式,我们平衡了最大化元数据覆盖率和保持统计可靠性。样本数量的分布如图2所示。接下来,我们策划和组合元数据组合,将元数据映射到已建立的本体,并纠正拼写错误。在过去的17年里,元数据被人工输入miniX,导致了“智人”、“智人”、“人类”、“人类”等一系列元数据组合,加上额外的空格或制表符的拼写,以及物种或器官的不同同义词。所有字符串都被转换为正式的本体条目,这是元数据组合减少的最大原因。总体而言,保留了515个元数据组合,标本减少了23.3%,总数为119,783个。下一种类型的数据争用是校正唯一箱的强度值。在这里,我们首先组合了由单一独特代谢物最能代表的箱子。由于多种衍生化形式(有或没有氨基的三甲基硅基化)或由于超载色谱导致的保留时间指数计算不正确,在17年的过程中出现了这种双箱。为了获得每个样品中每种化合物的单一强度,我们优先从人口最多的箱中提取强度。如果没有检测到该箱,我们根据两个箱之间的平均强度比缩放第二密集箱的强度。总体而言,我们保留了16,616个与元数据组合相关的bin(773个具有已知化学结构的代谢物,以及15,843个未知代谢物)。
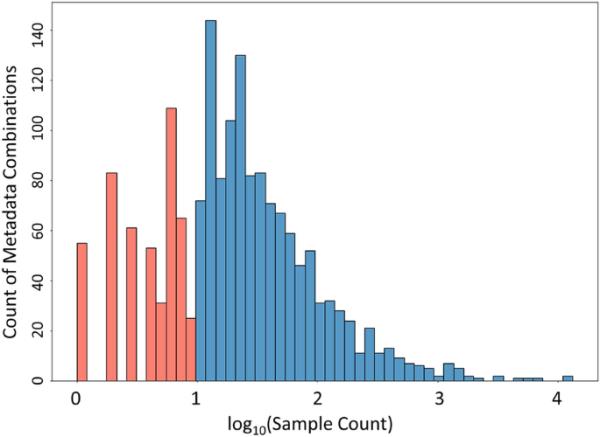
生物元数据三元组的所有组合的样本计数。为了提高统计可靠性,将少于10个样本的黑社会(红色)去除
最后,我们必须计算缺失的值。在这里,我们考虑了四种场景(附加文件3:图S2)。(1)特定的bin可能在样本中确实不存在,甚至可能在完整的元数据组合中也不存在。事实上,由于生物学的原因,大多数生物标本中没有大多数箱子。然而,当计算器官或物种之间的仓强度比时,当化合物在一个器官或物种中不存在而在另一个器官或物种中存在时,比值倍数变化将变得无限。(2)另一方面,由于随机误差(如峰值检测算法中的阈值),一个bin可能不存在。例如,如前所述,我们的Binbase算法基于信号强度对频谱质量使用保守阈值。如果一个峰值没有达到加权点分数相似度阈值700,则不会声明在该样本中找到该样本,并在Binbase数据库中丢失该样本。手动调查或递归回填可能会发现这样的峰值,但这些方法不容易处理。我们把这样的峰称为随机缺失(MAR),而真正缺失的化合物(由于生物学原因)可以被认为是非随机缺失(MNAR)。(3)在特定的元数据组合中,大多数峰值不是100%被发现,或者是0%被检测到,即总是不存在,而是介于两者之间。对于使用样本向量或特征向量的MAR代谢组学数据,已被证明可以很好地为缺失数据输入最小强度[20]。但是,如果某个特定元数据组合基本上不存在一个bin(即很少检测到),那么单个异常值可能会严重膨胀整个分布。因此,我们计算了存在的百分比,乘以每个元数据组合检测到的峰值(bin)的最小值。这样,如果几乎所有的样本都有注释,那么我们只需估算最小值。如果几乎所有的样本都没有注释,那么我们将估算一个接近噪声水平的小数字,并将保留半定量的折叠变化。该方法还提供了一种解决方案,用于解决约50%的罕见但具有挑战性的病例,其中数据既不能清楚地代表MAR也不能代表MNAR病例。(4)最后,如果一个箱子完全不存在,则不存在最小值。在本例中,我们输入了一个值,使得任何0% MDC的平均值将出现在该化合物跨所有元数据组合的平均分布的左边缘(这样,差异分析将显示比0%的情况有所增加)。因此,对于所有箱和所有元数据组合,给出一个值,通常作为一个小噪声项。在对所有bin和所有元数据组合的分布进行归一化、输入和整理之后,我们计算了bin强度的衍生物,以便对代谢组范围的元数据组合进行比较和查询。在这里,我们计算强度值的平均值、中位数和比率,并将结果数据集存储在PostgreSQL BinDiscover数据库中。我们还对对数变换后的分布对进行了Welch t检验。由于已知代谢物值的典型非高斯分布现象,我们在这里选择了对数变换数据,而不是直接使用Welch t检验。存储折叠变化和显著性计算的结果,而不是存储底层分布,以便大大加快用户查询的实时查询结果返回。
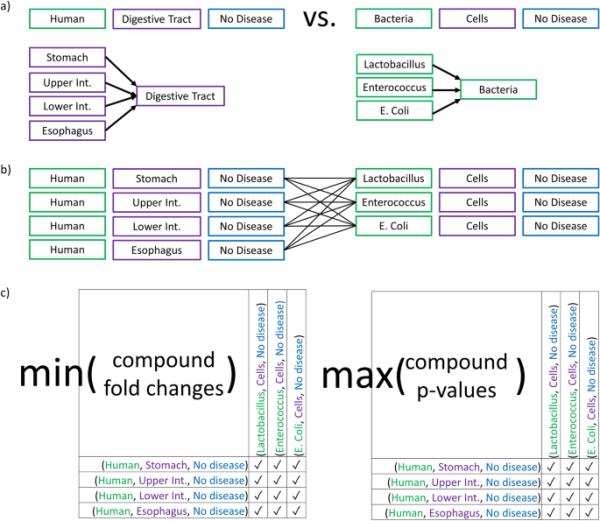
本体分组差异分析的模式。示例查询人类消化道与细菌代谢组。a将元数据在本体论上映射到(人类,无疾病消化系统)的所有Binbase样本与映射到(无疾病细菌细胞)的样本进行比较。b这种基于本体的摘要查询产生一组生物元数据组合,然后进行两两差分分析。c对于每种化合物,两两差分分析产生一个p值矩阵和一个折叠变化矩阵,它们可以分别用最大p值和最小折叠变化保守地描述。因此,在下游的火山地块中,每个化合物只能显示一个点
o病理分组差异分析
在这里,我们引入本体分组差异分析(OGDA)来提取隐藏在组学数据库中复杂数据中的泛化。在代谢组学以及蛋白质组学或基因组学数据库中,生物学家或生物医学科学家进行的研究包括复杂的研究设计,最终可以在与每个样本相关的生物元数据中进行描述。我们使用医学主题标题(MeSH)本体、ClassyFire化学本体和NCBI物种本体对可用的生物元数据进行了总结。因此,所有样本元数据都被表化为本体集。然后OGDA利用本体根据它们的分类接近度来选择集合。通过这种方式,来自许多研究的样本可以在数据库范围内的不同本体层次上进行比较。因此,难以处理的结果列表被转换为压缩列表,以进行进一步分析。
为了举例说明这种方法的强大功能,我们随机使用了三个用例,涉及跨物种的器官水平查询、跨物种查询、人类疾病水平查询和代谢物水平查询。图3演示了如何执行本体分组的差异分析计算。在这里,营养研究人员可能对查询人类消化道中发现的微生物细胞(细菌)和代谢物之间的代谢组学差异感兴趣。因此,示例查询将在非常通用的术语级别(消化道和细菌)上使用BinDiscover本体三元组[(人类,消化道,无疾病)vs.(细菌,细胞,无疾病)],而这些术语本身不会在研究元数据中找到。然而,这些词语和抽象在文献中被普遍使用和理解。
为了处理这个请求,BinDiscover将给定的请求转换为一个等效的请求,该请求利用了Binbase中所有相关的和可用的样本。本体搜索产生与“消化道”或“细菌”相关的所有样本,并获得与所请求的层次级别(“属于”)在本体上相关的所有节点的集合。详情载于附加文件3:表S1。重要的是,粪便(人类粪便)不属于消化系统的MeSH本体,而是属于“液体和分泌物”本体。因此,人类粪便样本不包括在这个特定的查询中。然后我们总结所有样本,并将更高本体级别的请求转换为相关元数据组合的列表。以这种方式汇总到查询组的所有Binbase样本的代谢组,然后进行两两统计分析。对于每一对,BinDiscover创建韦尔奇检验统计p值列表的经典结果以及两个查询集之间相应的折叠变化。因此,如果我们对一个本体样本集有n个组合,对比较器样本集有m个组合,我们就会为每个代谢物产生n*m个fold-changes和p值。然后可以将结果重新考虑为每个化合物的n*m折叠变化矩阵和n*m p值矩阵(图3c)。
接下来,BinDiscover将这些复合矩阵简化为每个化合物的一个聚合p值和相关的折叠变化。为了提取整个数据库的总体趋势,我们保守地估计了所有n*m对中每个化合物的结果。例如,如果至少有一种细菌的代谢物水平明显高于任何人类肠道器官,但其他细菌的代谢物水平不显著,则该代谢物不会被总结为细菌代谢与人类肠道样本之间的总体显著差异。因此,为了保持这种保守约束水平,我们使用每个化合物的最大p值和最小折叠变化作为边界。如果统计检验总体显著,但n*m对同时显示正折和负折变化,BinDiscover表示折变化为0。对于图3所示的示例查询,我们最终没有发现任何化学鉴定的细菌代谢物与人类肠道代谢组中检测到的显著不同或水平更高。然而,该查询检索到15种显著代谢物,发现它们在人体消化系统器官中水平升高(附加文件3:表S2)。这些化合物可归纳为维生素、脂类、固醇和氨基酸衍生物。这些代谢物确实不是由细菌直接产生的,但在广义上与人类的食物代谢有关,这证实了BinDiscover查询与可以从科学文献中获得的经典信息相匹配的有效性。当我们对773种结构上已确定的化合物进行测试时,我们在26秒内获得结果,每个元数据组合查询的速度大约为1秒。当我们对15,843种未知化合物重复分析时,BinDiscover在8分钟40秒内检索到结果,每次查询的速度为6.9秒。总的来说,我们发现74种未知化合物在细菌中含量明显较高,0种化合物在人体器官中含量较高。
案例研究1:探索食物tabolomes
代谢组学是一种假设生成工具。数据库必须通过提供特定查询来证明它们的有用性。我们在这里提供了四个用例,以突出生物学家或生物医学科学家如何使用BinDiscover网络工具。为了能够快速探索物种、器官和疾病之间差异的代谢组数据,用户可以在生物元数据上定义本体论分组的差异分析,或者从以化合物为中心的池中探索数据。该网络工具依靠普遍接受的统计数据和清晰的图形来快速了解生物比较中的主要代谢差异(图4)。
我们首先设想了一个营养研究人员探索这个工具。食物代谢组和饮食生物标志物越来越被认为是疾病的重要因素[21,22]。首先,研究人员可能会想知道为什么“一天一个苹果,医生远离我”?用户可能会选择将苹果与任何其他水果进行比较,在本例中是无花果(图4a)。这样的比较是有效的,并产生了大量的信息比较这两种水果。将鼠标悬停在在线图上(图4a)时,每个点代表一个单独的化合物。塔格糖在这里被突出显示为代谢物,在苹果和无花果果实中显示出最大的差异。此时,用户可能希望增加查询并将苹果水果与BinDiscover数据库中的所有水果(目前为26种水果)进行比较。通过这种方式,研究人员发现与所有其他水果相比,苹果中哪些代谢物独有地增加或减少。有趣的是,该查询仍然显示苹果中的塔格糖含量高于其他水果(图4b),与苹果/无花果对的差异分析相比,总代谢差异明显较小。与可视化图表相对应的在线数据表显示了所有差异代谢物,并指导用户进行特定于化合物的后续查询。在这里,设想的营养学家用户将找到一个日晒图和化学元数据(图4c, d)。日晒图显示,在所有物种/器官/疾病元数据组合中,塔格糖在苹果果实中显示出最高的强度。这一发现可能很有趣,因为塔格糖尽管含有蔗糖92%的甜度,却只提供蔗糖约1/3的卡路里[23]。此外,塔格糖不会增加2型糖尿病患者的胰岛素[24]。研究人员可能会将这一发现作为进一步研究的起点,例如,利用苹果基因组工具来增加其他水果甚至苹果品种中塔格糖的含量。
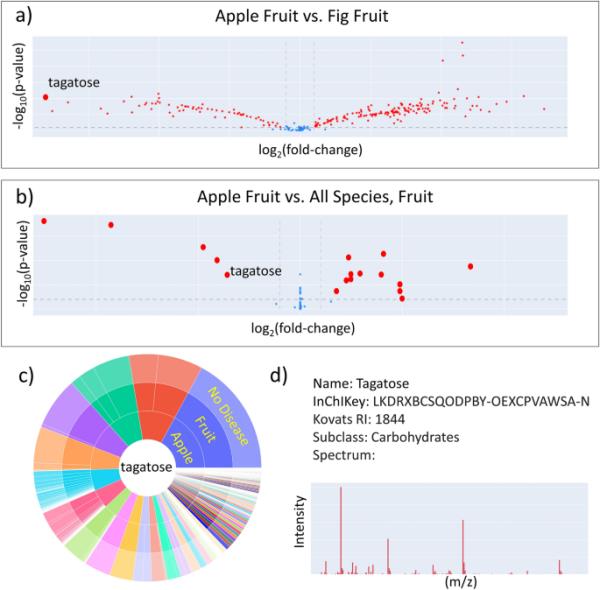
BinDiscover中的查询提供了新的生物学见解。a比较两个不同物种的特定器官的代谢组,例如:苹果和无花果,会产生许多差异。b将特定器官(苹果果实)与所有物种的相同器官进行比较,将总体差异限制在少数代谢物上。c然后查询了一种不同的苹果代谢物,塔格糖,发现与代谢组数据库中所有其他物种/器官组合相比,苹果果实中最丰富。然后给出塔格糖的化学信息,如质谱、定量质量、国际化学标识符、保留指数和化学类本体
案例研究2 -癌症我tabolism
接下来,我们设想一个对使用BinDiscover感兴趣的癌症生物学家。在这里,我们强调了如何重复利用BinDiscover差异分析工具来分离已识别的和未知的化合物,这些化合物将癌症代谢表型与相应的非恶性类似物区分开来,以及不同的癌细胞和组织如何揭示在其他癌症中不普遍的特定改变。具体来说,为了证明原理,我们获得了肺癌、肝癌和胰腺癌的三种元数据组合,并将其与非恶性肿瘤相比较。在每一次利用中,我们都得到了一组化合物。通过取结果集的交集,癌症生物学家可能会发现在所有癌症类型中受差异调节的化合物(图5a),以及每种癌症类型特有的化合物。我们发现了11种与所有癌症相关的化合物,如谷氨酰胺、脱水谷氨酰胺、n-乙酰谷氨酸和甲基丙二酸的增加(附加文件1:数据S1)。这些化合物可以与三羧酸(TCA)循环活性有关,特别是对于取代碳进入TCA循环的倒转反应。例如,已知过量的谷氨酰胺在癌细胞中大量使用,特别是通过谷氨酰胺脱氢酶产生谷氨酸,然后将其转化为α -酮戊二酸盐[25]。类似地,由于丙酮酸脱氢酶活性降低,癌细胞被剥夺线粒体乙酰辅酶a,支链氨基酸降解产物甲基丙二酸在倒转反应中转化为TCA代谢物琥珀酰辅酶a。BinDiscover联合差异分析发现的另一个典型的癌症生物标志物是焦磷酸盐增加,它与激酶活性增加和细胞生长有关[26]。此外,我们探索了可能区分这里研究的三种癌症类型的明显化合物(附加文件1:数据S1)。
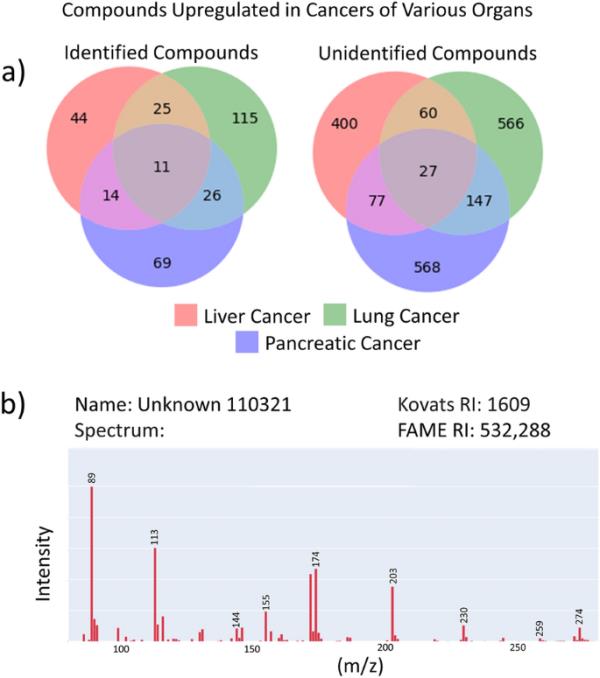
顺序查询提取与癌症代谢相关的未知代谢物。整合三个BinDiscover查询的结果,比较肝癌、肺癌和胰腺癌有癌症和没有癌症的研究,得到三组化合物。结果在这里分为已识别的和未知的化合物。b BinDiscover提供光谱和化学元数据,使化学家能够在自己的研究中利用未知化合物,无论是在自己的研究中靶向这些化合物还是用于化合物鉴定。这里显示unknown 110,321。
例如,在胰腺癌中,我们观察到所有四种形式的生育酚(也称为维生素E)的含量增加,维生素E被认为与降低胰腺癌风险有关,这与我们的研究结果相反[27]。我们还注意到一些二肽在胰腺癌研究中特异性增加,如胱氨酸、同型半胱氨酸和dialanine(附加文件1:数据S1),表明肽作为补充营养素的进口增强或蛋白质水解增加。在肺癌研究中,我们注意到α -酮酸,如2-酮异己酸和2-酮异戊酸,以及相应的α -羟基酸,如2-羟基戊酸和2-羟基戊二酸的水平升高(附加文件1:数据S1)。这些化合物通常与氨基酸降解的增加有关。肺癌研究也以乙酰化升高为标志,包括n -乙酰基甘氨酸、-甘露糖胺、-丝氨酸、-天冬氨酸和-腐胺(附加文件1:数据S1)。后两种化合物先前被认为是肺癌进展的生物标志物[28,29]。对于肝癌,在前列腺癌或肺癌研究中缺失的最明显的特定趋势是糖酵解中间体半乳糖-6-磷酸、果糖-6-磷酸、果糖-1,6-二磷酸、3-磷酸甘油酸、2-磷酸甘油酸和磷酸烯醇丙酮酸的丰富,以及戊糖磷酸循环代谢物核酮糖-5-磷酸和通用磷酸糖肌醇-4-单磷酸和n -乙酰氨基葡萄糖-6-磷酸(附加文件1:数据S1)。糖酵解通量的增加不仅在肝细胞中是众所周知的[30],而且也是癌症的一个普遍标志,根据Binbase的研究,与肺癌或胰腺癌相比,肝癌的糖酵解通量要高得多。除了经典的已知代谢物外,化学家和代谢组学研究人员可能会帮助癌症研究人员找到癌症代谢失调的新线索。在这里,我们发现了1500多种针对这三种癌症类型的未知化合物,以及27种在所有病例中普遍存在差异调节的未知化合物(附加文件2:数据S2)。图5b显示了从27种常见失调化合物中随机选择的一个例子的化学元数据,未知的110,321。由于Binbase同时提供了光谱、定量离子和保留指数,其他代谢组学研究人员可以很容易地利用这些信息在他们的研究中针对这些未知的癌症生物标志物。其次,新的生物标记物光谱作为化合物鉴定的起点。化合物110,321显示了一系列的偶离子片段,如m/z 144, 172, 174,这是典型的伯胺,加上m/z 274和m/z 230周围的高m/z离子簇,这也表明氮部分的存在。该光谱缺少m/z 117,这是羧酸和糖的典型片段。保留指数表明该化合物具有与其他氨基酸相似的沸点,因此,化合物110,321可归类为具有附加官能团(如仲胺)的伯胺。通过化学电离/精确质谱分析,可以识别出完整的结构[31]。
案例研究3 -细菌的多样性tabolism
微生物学家可能会使用BinDiscover来研究跨物种的细菌代谢,例如,作为合成生物学取代传统合成途径的背景[32]。同样,肠道微生物组作为许多内源性代谢物的来源以及药物检测中表型的起源也越来越受到关注[33]。细菌代谢功能潜力的多样性是我们感兴趣的,因此我们在BinDiscover中生成了一个聚类热图作为系统代谢组学工具(图6)。这些系统代谢组学热图利用所有检测到的代谢物(列)的化学分类存在,通过分层聚类(行)对分类群的指定组合进行分类。
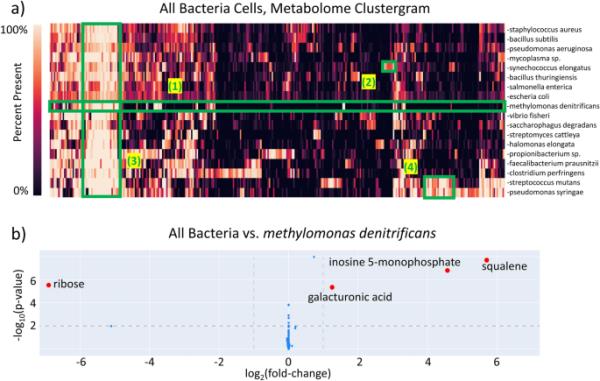
BinDiscover细菌的气相色谱代谢组比较。a BinDiscover中所有代谢产物对抗所有细菌种类的热图。基质进入颜色是由该物种中该代谢物存在的百分比决定的。四个感兴趣的区域(1)-(4)以绿色突出显示,并在文本中进行了讨论。b细菌种类与甲烷代谢物种甲基单胞菌代谢组丰度的差异比较
这样的热图可以用来描绘特定的异常物种,如图6a中突出显示的反硝化甲基单胞菌#1部分所示,它以甲烷代谢为碳源。BinDiscover对该物种与所有其他细菌的详细比较(图6b)显示,角鲨烯[34]和肌苷-5-磷酸的产量大大增加,同时减少了核糖的生物合成。图6a中的第2部分突出显示了长聚球菌(一种蓝绿色光合藻类)特有的一簇化合物,它除了产生色素反式叶绿醇之外,还产生各种烷烃,这些烷烃在BinDiscover中所有其他细菌中都不存在。第3部分含有普遍存在的代谢物,如脂肪酸、氨基酸和核酸,因此不能用于细菌分类。最后,第4部分标记了人类口腔细菌变形链球菌和植物病原体丁香假单胞菌之间的代谢物。观察到的代谢物包括色胺和吲哚-3-醋酸酯,这些已被纳入研究宿主-病原体关系的出版物[35,36]。总的来说,这些细菌代谢组的多样性反映了预期的生态位[37]。在这里介绍的案例研究中,我们主要关注生物学概念,因为BinDiscover本身本质上是面向信息学的。另一个案例研究更倾向于化学信息学,我们展示了未知化合物的相关性,如附加文件3:图S4所示。
目录
摘要 介绍 方法 结果 讨论 结论 数据可用性 缩写 参考文献 致谢 作者信息 道德声明 补充信息 搜索 导航 #####讨论
BinDiscover有效地实现了代谢组学信息的快速荟萃分析,目的是为生物科学家提供易用性,重点关注代谢组学覆盖的能力和广度。然而,生物样本元数据的事后检索和协调是具有挑战性的。据我们所知,很少有能够正确绘制生物学研究设计的可用界面的例子,不仅涵盖物种和器官,还包括研究设计的治疗、时间过程或疾病表型维度。因此,关于样本元数据的两个最重要的问题是,在我们的miniX研究设计数据库中捕获生物学研究信息时使用的元数据术语不一致,以及遗漏细粒度的生物学研究设计细节。不一致的元数据术语描述了过去18年来,生物学家向加州大学戴维斯分校西海岸代谢组学中心发送研究报告时,对样本进行标记的非正式性。虽然对于领域专家来说,“C57BL/6”这样的词可能足以描述特定的小鼠野生型,但即使对于这个经典的例子,也存在不同的实验室菌株,例如Jackson实验室的小鼠B6J(或B6/J),以及来自其他实验室的类似菌株变体。从细胞类型到组织和器官的细粒度描述,在其他生物领域也是如此。与这种细节的遗漏密切相关的是难以捕捉生物学研究的本质,例如使用特定基因敲除或药物治疗。生物学家有时通过文本格式和样本列表提供信息,但通常使用特定领域的缩写词,难以在我们的GC/MS数据库中对2000个不同研究的多样性进行回顾性编译。值得一提的是,所提出的案例研究强调了BinDiscover的生物学应用,作为其作为信息学工具的内在本质的补充。当然,BinDiscover的应用范围超出了这里所介绍的,我们通过附加文件3(图S4)提供了基于化学信息学的GC/MS代谢组中未知的发现。
以编程方式捕获研究设计细节的另一种方法可能是使用命名实体识别与NoSQL/GraphDB记录相结合。实体识别系统可能从已知本体的词汇表开始,但需要能够扩展内部一致的词汇表以捕获任意描述。虽然图形方法允许对样本及其关系进行鲁棒和动态描述,但命名实体识别避免了有问题的管理。然而,基于图形的界面会给用户带来很大的复杂性,特别是要求提交研究信息的生物学家。最初的努力导致了挫折,并淹没了潜在用户。捕获研究元数据的另一种方法是预先定义研究设计的主题,并将研究设计细节强制纳入这些主题。虽然许多细粒度的研究细节(因此,样本元数据分辨率)在粗糙的基于图案的GUI形式中丢失,但这些工具可以极大地简化生物学客户机的过程。虽然不全面,但减轻研究人员的负担可以极大地增加个人在使用代谢组学(或其他组学)服务时提供这些细节的可能性。
重要的是,本体论分组差异分析提供了重要的定量结果,而简单的存在/缺失分析忽略了这一点。例如,在人类血液中,通过非靶向GC-MS代谢组学可以检测到少量的蔗糖。然而,与植物样本相比,得出蔗糖是人体样本主要成分的结论是错误的。在这里,基于GC-MS代谢组学的半定量评估是可能的,有两个原因:(a) 70 eV的电子电离在GC-MS中标准化了60年,并且不受共洗脱化合物的抑制,不像LC-MS /MS使用的电喷雾过程。这条规则的一个例外是超过峰值饱和的化合物附近,例如尿中的尿素。(b)加州大学戴维斯分校的提取、衍生化、进样、检测和数据处理方法已经标准化,以确保色谱图不会过载(即避免峰饱和),但也不会空白(确保特定样品中最丰富的峰达到检测器饱和)。因此,通过数据采集和数据处理程序,包括在过去18年中使用完全相同浓度的(脂肪酸甲酯)内部标准品,可以保证半定量。然而,当然,尽管有这些预防措施,定量结果必须谨慎解释。例如,跨器官的比较可能包括生物流体与组织,即不同的生物量单位。此外,不同组织或生物流体的不同溶剂萃取效率可能会产生偏差。最后,当检测器在分离非常高丰度和非常低丰度的化合物时发生饱和时,丰度可能被低估。因此,产生较大倍数变化差异的定量比较比产生较小差异的定量比较具有更高的置信度。事实上,本体论分组差异分析的目标之一是保守地减少在所要求的器官中每种化合物的折叠变化,以增加对这些定量结果的信心。然而,对于样本和研究较少的生物元数据组合,定量比较不如Binbase中有数千个样本数据可用的差异分析可靠。
此外,BinDiscover是基于2021年12月的Binbase数据快照构建的。随着Binbase的不断扩展,新的化合物被添加进来。例如,在2022年11月,我们在一项分析牛肌肉组织的研究中首次可靠地检测到羧甲基半胱氨酸的存在(附加文件3:图S3),该研究使用抗氧化磷酸化复合物的抑制剂进行处理。Binbase中的化合物(如羧甲基半胱氨酸)在成功诱导到Binbase之前可能很少或丰度很低。为了克服这种元数据不一致,BinDiscover专注于使用本体论差异分析、太阳爆发图和种代谢组树对物种和器官查询进行高级分析。用户获得每个查询和元数据组合的样本数量,并且认为比较中包含的样本越多,对中位代谢物水平的估计就越可靠。如果用户关注具有足够大的折叠变化的化合物,甚至更具体的元数据比较可能提供对代谢差异的见解。
BinDiscover的目标是假设生成和数据探索。我们被激励去发现意想不到的发现,并且,在上下文中,相对不关心假阳性(类型I错误)。我们确实注意到,使用Benjamini-Hochberg程序或类似的方法来降低错误率是很常见的,但理性地避免了这一步,因为我们对增加可探索结果的召回感兴趣。同样,我们希望探索在探索数据本身的同时发现广义样本组的方法,因此我们在这里引入本体论分组差分分析。同样,在组合元数据组合时,我们不使用Fisher的方法来汇总p值,因为我们在每个两两比较中比较完全不同的假设,使用本体论分组差异分析。
BinDiscover的未来版本可能会根据来自新研究的数据(包括来自公众贡献的数据)进行增量更新。一个相关的工具,ADAP-KDB,从代谢组学工作台永久检索和更新用户可探索的光谱共识库[38]。dap - kdb不使用静态快照,并专注于社区贡献的数据源,但它显然是以频谱为中心的,并得到GC-MS中实际标准的辅助。我们希望社区范围内的努力能够进一步标准化元数据定义、样本提取、数据采集和数据处理的标准操作程序,从而自信地将社区更广泛的贡献纳入GC-Binbase。
代谢组学的荟萃分析系统关注样本,而不是研究,这一点至关重要。通过这种方式,样本的元数据可以重新用于从分析样本库进行新的生物学比较。目前,荟萃分析通常依赖于将具有大致相同意图的研究结合起来,这大大减少了数据可重复使用的方式。作为代谢组学数据大统一的一部分,我们希望代谢组学的标准化将得到改善。在提取前将内部标准试剂盒作为基质尖峰纳入样品中,可以作为仪器状态的检查,也可以进行半定量的实时校准,这将大大提高样品到样品集成的置信度。
结论
BinDiscover是一个基于156,000个样本GC-TOF数据库的web工具,该数据库自2005年以来积累了数据。我们通过删除未通过质量控制检查的样本,输入缺失值,并将元数据以及已识别的代谢物映射到已建立的本体来管理该数据集。我们展示了我们的webtool能够快速生成假设和趋势提取,以便将机器大小的数据库转换为人类大小的、可操作的简化。我们的工具提供了能够同时检查大量数据的组件,以及专注于单个化合物的能力。我们使用化学分类树和本体论分组的差异分析来实现多种类型的物种和器官的比较,并且还使用sunburst图或化学元数据对单个化合物进行可视化。一种新的数据分析方法,本体分组差异分析,使用外部本体,如NCBI物种分类法或MeSH层次结构,来创建匹配通用术语的样本组。本体分组差异分析的逻辑可以应用于任意元数据或特征,只要存在相应的本体,因此我们认为它也适用于其他组学。因此,可以沿着本体轴对查询进行分组,例如,将“啮齿动物的血”与“人类的血”或类似的广泛分组进行比较。代谢组学现在已经足够成熟,可以重新使用存储在标准化方法衍生的大型数据库中的数据,其明确目标是在不同的研究中进行荟萃分析。我们强烈强调代谢组标准化倡议的重要性,这对于交叉研究和跨物种数据比较至关重要。事实上,这种以样本为中心的数据收集可以形成大规模表型预测机器学习模型的训练集。我们发现,在创建这个元分析工具时,最具挑战性的一个方面是管理和协调生物学家客户提交的大量元数据。我们设想朝着简化但功能强大的元数据捕获系统努力。
补充信息
AdditioNal文件
确定了在癌症中上调的化合物。
AdditioNal文件2。
未知化合物在癌症中被上调。
AdditioNal文件
补充数字和表格。
下载原文档:https://link.springer.com/content/pdf/10.1186/s13321-023-00734-8.pdf


