


摘要
背景
同一菌种下的菌株可以表现出不同的生物学特性,因此菌株水平的组成分析是了解微生物群落动态的重要步骤。宏基因组测序已成为探索宿主相关或环境样品中微生物组成的主要手段。虽然有大量的成分分析工具,但它们并没有优化以解决菌株水平分析的挑战:高度相似的菌株基因组和样品中一个物种下存在多个菌株。因此,本工作旨在为短读段提供高分辨率和更准确的应变水平分析工具。
结果
在这项工作中,我们提出了一种新的应变级成分分析工具,名为strain scan,它采用了一种新的基于树的k-mers索引结构,在应变识别精度和计算复杂性之间取得了平衡。我们在大量模拟和真实测序数据上对StrainScan进行了广泛的测试,并使用流行的菌株水平分析工具(包括Krakenuniq, StrainSeeker, Pathoscope2, Sigma, StrainGE和StrainEst)对StrainScan进行了基准测试。结果表明,StrainScan在应变级成分分析上具有比现有工具更高的精度和分辨率。在菌株水平上识别多个菌株的F1分数提高了20%。
结论
通过使用新颖的k-mer索引结构,strain scan能够提供比现有工具更高分辨率的应变级分析,使其能够在一个样品或多个样品中返回更多信息的应变组成分析。StrainScan接受短读数和一组参考应变作为输入,其源代码可在https://github.com/liaoherui/StrainScan免费获得。
视频摘要
背景
越来越多的证据表明,由于基因组变异,一个物种内的菌株可能具有不同的代谢和功能多功能性[1,2,3]。同一种下的菌株可以表现出较高的序列多样性和不同的基因组织[4]。一个菌株的独特基因或snp可能导致新的酶功能、抗生素耐药性、毒力、不同的感染病毒等。例如,至少有数千种大肠杆菌菌株被鉴定出来,其中一些含有毒力因子,而另一些则是共生的。一个值得注意的例子是2011年德国爆发的大肠杆菌疫情,由菌株O104:H4引起,该菌株获得了编码志贺毒素的前噬菌体和其他毒力因子[5]。
由于不同的菌株具有不同的生物学特性,因此确定菌株对微生物组的组成和功能分析都具有重要意义。宏基因组测序数据包含来自宿主相关或环境样本的测序遗传物质,已成为研究菌株水平细菌组成的主要来源。越来越多的研究产生了关于菌株在不同样品中的基因型和表型的新知识。例如,Pollard等人发现,在198个海洋宏基因组中,许多流行的细菌物种具有与地理位置相关的菌株水平组成[6]。一项密切相关的研究表明,在克罗恩病患者的肠道微生物群中,优势大肠杆菌菌株会随着时间的推移而改变[7]。copri是人类肠道中另一种非常常见的细菌,已被证明其菌株与宿主的地理位置和饮食习惯有密切联系[8,9]。一些潜在的益生菌嗜粘杆菌菌株被发现具有抗炎特性,这可能对肥胖和糖尿病有有益作用[10]。此外,菌株在人体不同部位的分布也存在差异。例如,过去的一项研究[11]发现,从身体不同部位收集的痤疮C.和表皮S.菌株是异质的和多品种的。
StrainScan通过在已识别的簇内搜索菌株来实现更高的菌株级分辨率。相比之下,像StrainGE和StrainEst这样的集群级工具只返回已识别集群的代表性菌株,而不会搜索集群中的其他菌株。“S1”和“S2”是两个输入宏基因组样本。含有216株大肠杆菌菌株的真实集群(命名为集群1)菌株之间的B-C基因含量差异。B中的10株菌株共有1722个菌株特异性基因。C中的“GCF_001695515”和“GCF_013167975”分别是簇1中最长和最短的应变
尽管品系水平的分析很重要,但在种水平以下的分类分析仍然很困难。一个挑战来自于一个样品中可能同时存在多个高度相似的菌株[12]。例如,最近的一项研究[13]发现,2或3株表皮葡萄球菌可在人类粪便样本中共存,其Mash距离[14]约为0.005。同样,有报道显示,作为人体皮肤微生物组的重要组成部分,痤疮C.的多株菌株经常形成复杂的混合物[15]。其中一些共存菌株表现出很高的序列相似性,其Mash距离约为0.0004。此外,一项分析2144个人类粪便宏基因组的研究[16]显示,许多样本含有高度相似的多氏拟杆菌菌株,它们彼此共存。常用的宏基因组分类和组装工具并不是为了区分不同的菌株而设计的。虽然有菌株分析工具,但它们可能需要来自同一种群的多个样本[17],只输出优势菌株[18,19,20,21],或者对菌株之间的相似性有限制[22]。紧接着的第二个挑战是菌株水平鉴定的解决方案。这里的分辨率体现在参考数据库的大小上,参考菌株的数量越多,分辨率越高[23]。虽然有些菌株具有很高的相似性,但没有已知的相似性截止点,低于该截止点,遗传差异就可以忽略不计。如致病性大肠杆菌CFT073和益生菌大肠杆菌Nissle 1917的序列相似性为99.98%[24]。同样,一项噬菌体-宿主协同进化研究[25]发现,即使菌株基因组ANI较高(>99.9%),菌株也可以被不同的噬菌体感染,表现出不同的防御或吸附机制。对于一些具有高品系多样性的物种,即使少数snv也会导致表型变异[26,27]。因此,更高的分辨率可以更准确地表征基因型和表型之间的关系。stringe[28]和StrainEst[29]等工具旨在解开菌株混合物的缠结,但仅限于在样本菌株基因组数据库中报告具有代表性的菌株。他们的聚类截止点(0.9 k-mer Jaccard相似性(strain)或99.4% ANI (strain))仍然可以导致一些细菌的大聚类。虽然StrainGE可以进一步识别样品中已识别的代表性菌株的snp /缺失,但它不能在已识别的集群中精确定位特定菌株。Krakenuniq[30]和StrainSeeker[31]这两种基于k-mer的工具在菌株水平鉴定中,当数据库中的菌株具有很高的相似性时,分辨率也很低。第三个挑战是鉴定低丰度菌株。例如,de novo应变构建工具[32,33]旨在通过基于装配的策略重建应变,通常需要较高的应变覆盖率才能实现准确的应变重建。此外,许多应变分析工具[34,35,36]也要求应变覆盖率大于10倍才能得到准确的鉴定结果。因此,识别这些工具覆盖率低的菌株仍然是一个挑战。最后一个挑战是应变识别时间。根据最近发表的研究[23,37],当数据库较大时,大多数基于校准的菌株水平识别工具(包括Sigma[38]和Pathoscope2[39])在计算上可能会很昂贵。大型参考数据库虽然可以增加物种内多样性的覆盖范围,但也需要更多的计算资源。
因此,迫切需要为宏基因组数据提供更灵敏、准确和高效的菌株水平分析。在这项工作中,我们介绍了StrainScan,这是一个开源工具,可以准确地从测序数据中检测已知菌株,包括宏基因组数据或全基因组测序数据。为了在分辨率和计算复杂性之间取得平衡,我们开发了一种新的分层k-mers索引结构,用于大量菌株,这些菌株通常表现出异质相似性分布。在第一步中,我们将高度相似的菌株聚类成簇。然后,我们设计了一种新的聚类搜索树(CST),一种基于树的聚类搜索索引结构。通过仔细平衡每个节点上k-mers的数量,我们优化了CST,以防止对低丰度菌株的假阳性菌株识别。在第二步中,我们使用菌株特异性k-mers和代表snv和结构变化的k-mers来确定哪些菌株可能存在。菌株扫描的最终输出包括鉴定的菌株及其丰度。通过在识别的簇内搜索菌株,strain scan比诸如StrainGE和StrainEst等簇级工具获得更高的分辨率,这些工具在每个簇中只保留一个代表性菌株。如图1A所示,不同的分辨率会导致不同的观察结果和结论。虽然StrainScan可以识别样品S1中的两种不同的菌株,但StrainGE或StrainEst不能区分它们,因为它们来自同一簇。同样,在比较两个样本(S1和S2)时,确定一个特定的菌株而不是一个簇,可以导致更准确的基于基因组成的分析,因为一个簇中的菌株仍然可以拥有非常不同的基因含量(图1B和C)。
通过将StrainScan与其他可用工具在多个模拟和真实测序数据集上进行基准测试,我们证明StrainScan可以比最先进的工具以更高的精度输出应变级组成。特别是,与stringe等最先进的工具相比,strain scan在识别应变水平的多个应变方面将F1分数提高了20%以上。strain scan是一种目标菌株组成分析工具,需要用户提供感兴趣细菌的参考基因组。通过支持自定义构建任意参考基因组的索引结构,它可以应用于任何细菌。
方法
StrainScan概述
StrainScan设计用于直接从短读数中识别已知菌株。由于有许多种水平的元基因组数据组成分析工具,因此strain scan的输入是“fastq”格式的短读数和“fasta”格式的目标细菌的菌株基因组。为了在应变识别分辨率和计算成本之间取得平衡,我们设计了一种分层索引方法,该方法结合了快速但粗粒度的集群搜索树(CST)和慢速但细粒度的集群内应变识别策略。如图2的流程图所示,我们首先创建一个基于簇树的索引结构。使用我们在这棵树上高效而准确的聚类搜索方法,我们可以首先确定样本中存在的一个聚类。然后,我们将使用精心选择的k-mers来区分鉴定集群中的不同菌株。分层方法有几个优点。首先,它允许我们适应菌株之间的异质相似性分布,一些菌株的相似性比其他菌株高得多。我们的快速CST搜索策略可以快速识别出相似度较低的菌株。只有那些高度相似的菌株才需要在第二步中进行更精细的区分。其次,分层方法可以通过允许我们使用更多唯一的k-mer来提高搜索精度(补充表S1)。现在集群之间共享的任何k-mers都可以用于集群内搜索。第三,分层方法可以减少内存占用。如果没有分层方法,我们需要从包含大量k-mers的所有参考文献中搜索菌株。对于CST搜索识别的簇,StrainScan只需要在识别的簇中搜索包含较少菌株和k-mers的菌株。例如,聚类前大肠杆菌参考集中的k-mers总数为192,325,016,而聚类后最大聚类的k-mers数量减少为16,071,080(补充表S1)。
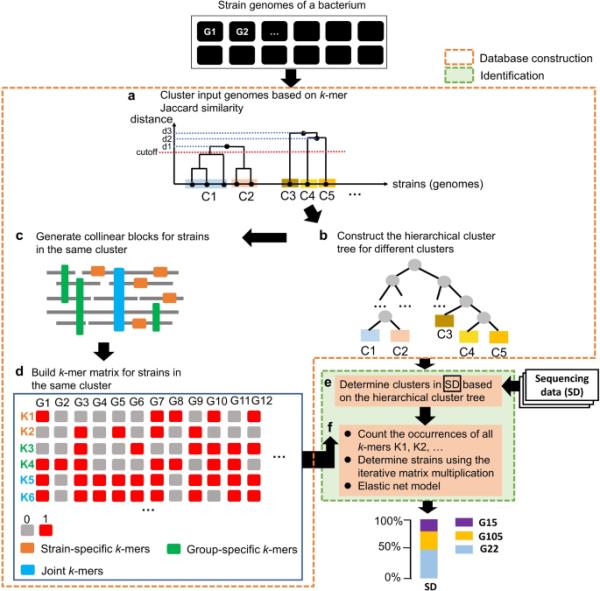
StrainScan的概述。(a)菌株基因组聚类过程示意图。给定感兴趣细菌的菌株基因组(G1, G2,…),所有对所有k-mers Jaccard相似度使用邵氏法计算[40]。然后使用单链接分层聚类对基因组进行聚类。默认情况下,聚类阈值设置为Jaccard相似度0.95。在本例中,给定红色虚线表示的截止点,聚类过程输出从C1到C5的5个聚类。(b)给定聚类,构建分层聚类树,用于以后的聚类级别识别。(c)生成共线块,提取有助于区分同一簇内不同菌株的k-mers。(d)步骤d总结参考基因组的索引结构过程。(e)和(f)为菌株搜索输入索引结构和测序数据(reads)。(e)搜索集群。(f)通过迭代矩阵乘法识别应变,最后通过弹性网回归推断出相对丰度剖面
一个基于cst的索引结构中k-mers赋值的例子。每个节点都具有其根子树所特有的k-mers,并且该子树中的大多数菌株共享k-mers。在本例中,每个带有特定颜色的条代表一个k-mers,每个节点被分配一个唯一的k-mers。B在构造节点v的k-mers集时,将所有叶节点分为三组,分别命名为、、和
聚类搜索树(CST)的构建
考虑到同一物种的许多菌株基因组,我们首先使用基于k-mers的无比对方法计算了Jaccard相似性矩阵[40]()。然后,我们在此矩阵的基础上进行了聚类层次聚类(单链接),将菌株分组成一个树状图。最后,我们选择了一个固定的高度截断H(默认为0.95),将树状图切割成许多簇,其中包含一个或多个菌株。每个簇内的菌株具有基于k-mer的Jaccard相似性,大致对应于99.89%的平均核苷酸同一性(ANI)[28]。
为了精确定位包含应变的簇,我们将簇和树状图转换为CST,以支持准确和高效的簇搜索。CST保持与树形图相同的树拓扑结构,除了每个集群由树中的叶子节点表示之外。此外,我们丢弃了树形图中的距离信息,以便每个节点与其父节点(或子节点)之间的距离是一致的,而不考虑它们的Jaccard相似性。因此,CST是一个全二叉树。为了支持集群搜索,每个节点包含一组k-mer,这些k-mer对于由该节点根的子树是唯一的。通过进行k-mers匹配,CST将引导我们取左子节点或右子节点,直到到达一个或多个叶节点(即簇)。我们首先描述如何为每个节点分配k-mers。
k-mers节点分配
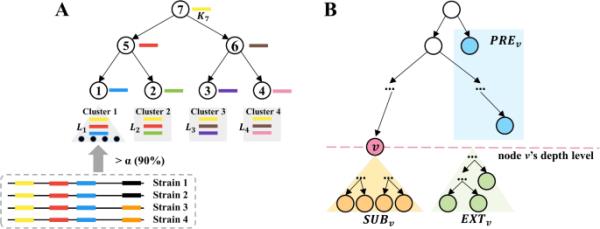
CST由两个元素定义:树拓扑和为每个节点分配的k-mers集。在本节中,我们将描述如何为节点分配k-mers以支持集群搜索。对于CST中的节点v,我们将以v为根的子树表示为。k-mers对v的赋值遵循两个标准。的叶节点上的大多数菌株应该共享k-mers。其次,k-mers是美国菌株独有的。使用图3A中的示例将这两个标准可视化。
根据这两个标准,我们首先用从菌株对应簇中提取的k-mers分配叶节点。为了使用在潜在菌株中代表相对保守性较好的特征的k-mers,对于具有多个菌株的集群,仅保留至少在菌株中出现的k-mers。大表示只有许多菌株共有的k-mers被用于构建CST,而小则允许CST使用菌株特异性k-mers。我们在实验中使用一系列的参数比较了聚类识别的性能。根据补充图S1的实证结果,我们将默认值设置为90。
因此,我们将叶节点v的初始k-mers集记为。接下来,从叶节点开始,我们递归地将每两个兄弟节点之间的共享k-mer移向它们的父节点。在最后一步中,所有出现在多个节点中的k-mers都将被删除。在此过程结束时,每个节点v(内部节点或叶节点)包含一组唯一的k-mer,记为。具体而言,对于节点v,可以使用如式(1)所示的集合运算来构造。对于具有深度的节点v,根据与v的关系将所有叶节点分为三组,如图3B所示,定义如下。
 (1)
(1)
目前构建的CST类似于StrainSeeker[31]中构建的树。虽然使用唯一k-mers可以指导搜索识别菌株簇,但一个显著的限制是一些节点只包含少量唯一k-mers,这比具有许多k-mers的节点更容易导致假阳性(FP)匹配。这是在使用StrainSeeker时观察到的。以112株copri菌株构建的StrainSeeker数据库为例。在222个节点中,21个节点是空的,104个节点的k-mers小于1000。具有较小k-mers集的节点往往是偶然匹配的,从而导致FP识别。为了解决这个限制,我们将通过添加不会给集群搜索增加歧义的k-mers来增加这些节点。CST优化方法的细节可以在补充章节1.1中找到。
CST中的聚类搜索
给定输入的测序数据,我们首先从CST中提取所有k-mers,并使用Jellyfish对所有短读段进行快速k-mers匹配[41]。然后,所有读取的k-mers匹配计数将被映射回CST。每个节点v将被分配一个一维数值向量,每个单元记录一个k-mers匹配计数。聚类搜索算法基于广度优先搜索(BFS),从根开始逐级检查节点的k-mers匹配(图4)。每个节点v的k-mers匹配向量用于基于二项检验决定是否遍历v的后代。最终的搜索结果包含一个或多个叶节点,表示测序数据中存在的菌株簇。
评分标准
当搜索访问节点v时,将计算两个评分指标来决定访问哪些子节点。如图4所示,第一个指标是匹配k-mers的比例(),它代表了测序数据中存在的k-mers的比例。定义为:
(2)其中表示包含所有正k-mers计数的向量。
第二个指标是平均k-mers匹配计数(),它仅使用具有正匹配计数的k-mers计算。当,设为0。
(3)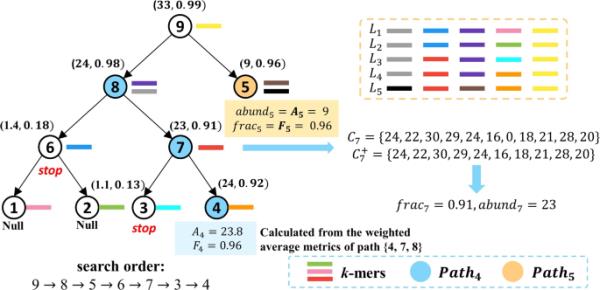
集群搜索过程的一个示例。两个评分指标的值显示在每个节点v旁边。搜索结果包含集群4和5,估计丰度分别为23.8和9。节点6和3没有通过二项检验,因此无法遍历它们的后代。和中的节点分别用蓝色和橙色表示。有相同的橙色k-mers。因此,我们需要根据集群5的估计丰度进行调整,以计算准确的评分指标
搜索策略
在计算出v的两个评分指标后,我们进行二项检验来确定CST中的遍历顺序[18]。由于测序错误可能导致k-mers匹配,二项检验的主要目标是区分测序错误的随机匹配和真实菌株的真实匹配,这对于低丰度菌株尤其重要。给定一个节点v,我们检查是否可以拒绝由排序错误产生的零假设。
具体来说,我们首先将和(p是v的父节点)四舍五入到最接近的整数和。然后,给定测序错误率e(默认为1%),当由测序错误产生的概率小于(默认为0.05)时,我们拒绝错误导致的零假设。概率估计为
(4)式中为二项分布的概率质量函数,包含试验次数和成功率。未能拒绝原假设表明我们无法区分低覆盖率k-mers匹配和测序噪声。因此,我们认为只是从排序错误,并停止搜索v的后代。否则,如果我们成功地拒绝零假设,我们认为在测序数据中存在一个或多个菌株簇。CST搜索将把v的两个子节点添加到BFS队列的末尾,准备稍后遍历它们。与传统的二叉搜索树(BST)不同,同一父节点的两个兄弟节点都可以拒绝错误导致的零假设。因此,我们可以遍历它们的所有后代,CST的搜索结果可能不止一个。
集群识别
一旦到达叶节点,我们将使用叶节点及其包含从叶节点移动的k-mers的祖先节点进一步检查k-mers匹配和丰度估计统计。如果只有一个叶子节点被识别,那么从根到v的路径上的所有节点都可以用来计算k-mers统计量。但是,如果标识了多个叶节点,则不应该使用所有的祖先节点。相反,应该只使用那些对叶节点v有唯一贡献的节点来计算最终的丰度。这些节点可以共同构成一条路径,其中所有匹配的k-mers仅来自叶节点v中的菌株。为了识别,我们首先识别只包含v的最大子树作为识别节点。和等于这个子树的根到v之间的路径。使用所有节点的k-mers计数来估计集群的丰度将比使用单个叶节点提供更高的置信度。以图4为例,识别出两个叶节点4和5。在本例中,是节点8的根子树中包含v的根到叶路径。随后,我们可以收集所有k-mers匹配计数,计算匹配k-mers的加权平均分数和加权平均k-mers匹配计数:
(5) (6)如果大于给定的截止值(默认值为0.4,但用户可以修改该值以适应不同的条件),我们认为v中的集群存在于测序数据中。在完成CST搜索后,将输出所有已识别的簇及其估计丰度(由)计算。此外,当测序数据中包含不同簇中的多个菌株时,由于弱节点扩增过程中增加了k-mers,会引入一些FPs。解决这个问题的详细方法可以在补充章节1.2中找到。
集群内的菌株鉴定
对CST进行了优化,以区分在给定截止点以下具有相似性的聚类。使用CST来区分高度相似的菌株可能会导致大量的弱节点,由于共享k-mers的百分比很大而无法增强。因此,一旦我们确定了一个集群,我们需要一种细粒度的方法来区分高度相似的菌株。一旦我们确定了一个簇,与原始问题空间相比,要区分的菌株数量大大减少。因此,我们可以使用所有具有区分力的k-mers。使用的第一个特征是来自菌株特异性区域的独特k-mer,这里我们称之为菌株特异性k-mer。使用的第二个特征是群体特异性k-mer,它可能来自某些菌株共同的结构变体(SVs)。在最近的一项研究[42]中,SVs被用来区分不同的菌株。受该研究的启发,我们从一些菌株共有的sv中提取了群体特异性k-mers。然而,在某些情况下,仅依靠菌株特异性和群体特异性k-mers仍然存在低分辨率问题。例如,在图5中,菌株4和菌株5都具有相同的群特异性k-mers,当样品中不存在菌株5的菌株特异性k-mers时,我们无法对两株进行精细区分。因此,为了进一步提高分辨率,我们添加了联合k-mers集,其中包含所有基因组中存在的核心基因组区域的snv和索引[29,38,43]。如图5所示,对于所有的关节k-mers,虽然每个k-mers不是特定于应变的,但每个应变设置的关节k-mers是唯一的。然而,关节k-mers的数量往往不如前两种k-mers多(补充表S2)。它们需要结合在一起,以提高识别的分辨率。利用这三种类型的k-mers,我们提高了识别的分辨率,同时减少了搜索空间。
.jpg)
使用菌株特异性k-mers、群体特异性k-mers和联合k-mers来区分同一簇中的5株菌株。每个菌株都有独特的k-mers组合
为了有效地提取这些k-mers,我们使用了Sibeliaz[44],这是一种高效的工具,用于识别密切相关基因组中的局部共线块。基于Sibeliaz生成的区块,我们开发了一种基于哈希的算法,从菌株基因组中提取这些k-mers,并将其保存在矩阵中以供以后使用。该算法的主要伪代码显示在补充章节1.3中。该算法的输入是Sibeliaz生成的同一簇和块中的菌株基因组。通过使用高效哈希表,该算法可以快速提取目标k-mers。最后,将从一个簇中提取的所有k-mers保存在一个大小为X的矩阵中,其中M为k-mers的数量,N为该矩阵中的菌株数量。如果应变j与k-mer相等,则为X,否则为X。当有多个簇时,分别生成多个对应的矩阵。
使用选择的应变识别k即
在上一步提取k-mers后,我们需要使用这些特征进行菌株识别。为了解开同一簇中紧密相关的复杂群落,我们采用迭代矩阵乘法来确定所有共存的菌株,并使用弹性网络回归预测它们的相对丰度。
迭代矩阵乘法的主要目标是通过使用k-mers矩阵中的三种类型的k-mers(图5)来确定同一簇中的菌株。为了实现这一目标,我们使用类似于QuantTB[37]的迭代策略,将样本中的k-mers与k-mers矩阵X中的k-mers进行比较。方法描述如下。给定树搜索选择的聚类和k-mers矩阵X中的k-mers,我们将使用Jellyfish[41]对测序数据中所有这些选择的k-mers进行计数。表示水母中所有选定k-mers的出现次数为向量y:,其中和表示矩阵中第i个k-mers的出现次数。然而,来自其他已识别簇的重叠k-mers可能导致错误的k-mers匹配或错误的丰度估计。为了消除其他聚类的影响,如果在树搜索检测到的其他聚类中发现了一个k-mers,则将其出现次数替换为0。对于k-mers矩阵X,其第j列X[:, j]定义为:
(7)基于X和y,我们使用迭代矩阵乘法,可以准确快速地检测样品中所有可能的菌株。给定X和y,该函数将为每个菌株计算一个分数X。注意,我们将超过第5百分位和第95百分位的值视为异常值,并将所有异常值设置为0。该功能将根据其得分对所有菌株进行排名。排序完成后,该函数将输出排名表中排名前1的菌株,然后通过将识别菌株中所有k-mers的出现次数替换为0来更新y。这个过程是重复的。它在每次迭代中继续计算分数并识别最可能的应变,直到具有非零值的k-mers的出现次数低于给定的阈值,其默认值为k-mers。本工作中所有实验均采用默认截止。
已知样品中可能存在的菌株,我们使用弹性网络回归模型来预测鉴定菌株的测序深度和相对丰度。我们选择弹性网模型而不是Lasso模型,因为Lasso模型倾向于低估应变的数量,导致召回率下降。经过迭代矩阵乘法,得到过滤后的k-mers矩阵X,其中为已识别菌株的个数。排序深度是回归系数,通过最小化弹性网惩罚残差平方和来预测:
(8)和是影响模型性能的两个重要参数,因此需要进行调优。我们设计了一个基于交叉验证的函数来调整和获得具有最低预测误差的模型。给出最佳模型,通过回归系数的归一化计算应变相对丰度。但是,如果通过树搜索检测到多个簇,则将根据簇的丰度重新计算一个菌株i的相对丰度。因此,各菌株i的最终相对丰度(RA)计算为:
(9)其中,C为树搜索预测的聚类丰度,n为所有已识别菌株的总数。
预测精度e估值
为了测试每种方法的性能,我们计算了每个测试类别的查全率、查准率和F1分数。真阳性(TP)是指正确鉴定出的菌株数量。假阴性(FN)是指样品中存在但被工具遗漏的菌株数量。假阳性(FP)是指错误鉴定菌株的数量。
在所有实验中,我们都使用Jensen-Shannon散度(JSD)[55]来测量真实的相对丰度与预测的相对丰度之间的距离。如果预测丰度和真实丰度具有不同的维度,我们将通过在具有较低维度的丰度上加零来计算JSD。假设存在两个概率分布T和P,它们的Jensen-Shannon散度介于[0,1]之间,定义为:
在哪里
称为从T到K的Kullback-Leibler散度,定义为:
目录
摘要 背景 方法 结果 讨论 结论 数据和材料的可用性 参考文献 致谢 作者信息 道德声明 补充信息 搜索 导航 #####结果
由于StrainScan专注于识别已知菌株,因此我们测试了StrainScan在六种细菌上的性能,这些细菌可能对菌株水平分析构成计算挑战。所有选定的细菌都至少有100个序列菌株。其中一些有大量已知菌株,如大肠杆菌和表皮葡萄球菌。有些菌株具有极高的序列相似性,如结核分枝杆菌。此外,我们选择了通常生活在不同生态系统中的细菌,如人类肠道和人类皮肤,包括嗜粘液杆菌、copri和痤疮杆菌。我们进行了多个实验来评估StrainScan。所有实验的总体情况如表1所示。首先,我们测试了strain scan在模拟数据和加标宏基因组数据中识别一个菌株和多个共存菌株的能力。我们通过配置应变相似度和应变测序深度等参数生成不同的数据集,帮助我们比较不同工具在困难场景下的性能。其次,我们在三个模拟社区数据集中测试了strain scan,这使我们能够在已知菌株组成的真实测序数据中评估不同的工具。第三,我们在94个不同深度的真实测序数据集上测试了StrainScan (Supplementary Table S3)[21,29,37,39,52]。因为在真实的测序数据中,通常没有菌株组成的基本真理,所以我们选择由数据作者分析过的数据集。通过对分析结果的比较,我们可以得出不同工具性能的一些结论。在这些实验中,我们使用F1分数、精度、召回率和Jensen-Shannon散度作为评估指标,这些指标在“方法”一节中定义。我们将StrainScan与流行的基于参考文献的菌株水平分析工具进行基准测试,包括Krakenuniq (V0.5.8)[30]、StrainSeeker (V1.5)[31]、Pathoscope2 (V2.0.6)[39]、Sigma (V1.0.1)[38]、StrainGE (V1.1.5)[28]和StrainEst (V1.2.4)[29]。
在这些测试工具中,StrainGE和strain将菌株分成簇,每个簇只保留一个有代表性的菌株[28,29]。因此,我们在两个分辨率水平上评估了它们的性能:菌株水平和集群水平。菌株水平评估只有在鉴定的代表性菌株与当前菌株相同时才将输出计数为真阳性(TP)。如果返回的代表性应变与目标应变在同一集群中,则集群级评估将输出计数为TP。相应地,在集群级别上,FP的定义也更为宽松。对于所有其他工具,我们使用应变级分辨率来计算相关统计数据。下面我们给出实验结果。
参考database建设
对于这项工作中测试的所有物种,我们尽可能全面地创建了参考菌株基因组数据库。因此,我们从NCBI RefSeq数据库中下载了测试细菌的所有完整基因组和草图。但是有25349个大肠杆菌基因组,需要超过1TB的内存。由于硬件资源的限制,我们只使用了来自RefSeq的完整大肠杆菌基因组。与大肠杆菌类似,我们的硬件资源使我们无法使用结核分枝杆菌的所有草图和完整基因组。此外,一些现有的结核分枝杆菌基因组仅相差不到10个位点[37]。这些几乎相同的菌株将在预处理步骤中聚集在一起。因此,我们使用dash[40]计算了所有结核分枝杆菌菌株的成对Jaccard相似性,并使用99%的k-mers Jaccard相似性阈值进行了完全连锁聚类。然后,我们只保留与该集群中所有其他基因组平均相似性最高的菌株。结果,6752个基因组中有792个被保留下来用于结核分枝杆菌。
菌株的最终数量及其其他特性记录在表2中。作为所有测试工具的输入的基因组数量显示在“输入基因组#”一栏中。如前所述,StrainEst和stringe将对输入的菌株基因组进行聚类,并在最终的数据库中只从每个聚类中选择一个具有代表性的菌株。结果,菌株数量明显减少(表2)。当我们仔细观察StrainEst和StrainGE的聚类时,我们可以发现,代表菌株与同一聚类中的其他菌株在基因含量和snv上存在显著差异(Supplementary Fig. S3, S4, S5)。对于某些物种,实际菌株与代表菌株之间的snv超过5000。在同一簇中,最长的菌株比最短的菌株具有1000多个基因。最近的一项研究[56]表明,在特定菌株中发现的“单基因”(unique genes)对于了解菌株特性非常重要。因此,在同一簇的菌株之间,这些较大的基因含量差异可能导致不同的特性和功能。一个值得注意的例子是,两个高度相似的菌株,e.c oli CFT073和e.c oli Nissle 1917,分别是致病性菌株和益生菌,通过StrainEst和StrainGE将它们归为同一簇。
在进行簇内菌株识别之前,strain scan还将菌株分组成簇。实验结果表明,使用CST的聚类搜索对所有被测细菌的准确率达到100%。对于大多数细菌,StrainScan具有比StrainEst和StrainGE更细粒度的簇(补充图S6),表明在簇水平上具有更高的分辨率。
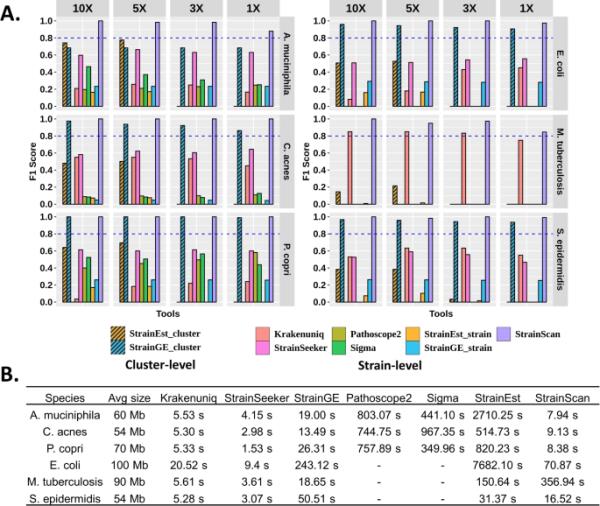
A不同测序深度下7种工具在“单菌株”模拟数据集上的F1得分。B 7种测试工具的运行时间比较。Sigma和Pathoscope2在一些数据集上没有价值,因为它们的计算成本太高,无法为相应的细菌构建数据库或从模拟读取中识别菌株
从模拟读数中检测参考应变
本实验的目的是测试strain scan和其他识别样品中当前菌株的工具。这里有两个挑战。第一个挑战是将真正的菌株与其他高度相似的菌株区分开来。第二个挑战是识别低深度的应变。因此,我们生成了多个具有不同测序深度的数据集。
对于每个细菌,我们随机选择一个参考菌株,并将其模拟的短读数作为所有工具的输入。为了避免任何与数据相关的偏差,我们重复实验60次,每次随机选择一个菌株。对于copri,我们只重复了50次实验,因为它的基因组数量很少。对于每个选择的菌株,我们模拟了不同测序深度(10X, 5X, 3X, 1X)的读数。因此,总共有1400个数据集。对于每个数据集,我们使用ART[57]模拟Illumina读取,参数如下:-p -l 250 -f depth -m 600 -s 150,其中depth为指定的测序深度。StrainScan和其他6个程序用于从这些模拟读数中识别菌株。由于Sigma和Pathoscope2的计算成本昂贵,我们无法构建大肠杆菌、表皮葡萄球菌和结核分枝杆菌的数据库。
各方案F1得分如图6A所示。TP、FN、FP、召回率和精密度记录在补充表S4中。在这些选择的菌株中,其中一些与至少一个其他参考菌株基因组具有99.5%以上的基于k-mer的Jaccard相似性。因此,一些工具的F1分数很低。当深度大于1X时,StrainScan在所有数据集上获得近乎完美的F1分数。然而,对于含有高度相似菌株的种(嗜粘杆菌和结核分枝杆菌),当深度为1X时,StrainScan的F1分数略有下降。当深度较低且应变相似度很高时,CST算法由于唯一覆盖率低而无法识别某些应变,召回率下降。尽管如此,StrainScan仍然对这些物种有最好的F1分数。目前,StrainScan可以接受的最小深度是1X,如果深度低于1X,性能会迅速下降。
虽然Pathoscope2和Sigma实现了相对较高的召回率,但它们的输出中有很多FPs,这使得它们的精度远低于其他工具。Krakenuniq在许多菌株具有独特k-mers的数据集上获得了更高的F1分数。然而,一些高度相似的细菌菌株导致了Krakenuniq的低召回率和准确性。StrainSeeker和StrainEst也有大量FPs,导致精度较低。此外,StrainEst无法识别深度低于5X的菌株。在许多数据集中,stringe在集群级分辨率上的表现与StrainScan相当。然而,当排序深度减小时,它返回更多的FPs。例如,当应变深度从10倍减小到1倍时,strain对C. acnes的簇级精度从0.95下降到0.76。相反,随着深度的减小,StrainScan不产生任何FPs (Supplementary Table S4)。即使在聚类水平上,菌株对结核分枝杆菌的表现也不理想,因为这些菌株具有很高的k-mers Jaccard相似性。在测试的工具中,StrainSeeker倾向于返回相同分数的多个菌株。这类似于通过StrainEst和stringe返回具有代表性的菌株,其中不提供一组菌株之间的更细微的区别。因此,这些工具的分辨率很低。例如,stringe返回大肠杆菌大小约为200的集群的代表性菌株(补充图S7)。根据我们之前对这些集群中菌株的遗传差异的分析(补充图S3),分辨率并不理想。
不同工具的运行时间如图6B所示。菌株扫描对除结核分枝杆菌外的所有测试细菌都有效。由于结核分枝杆菌菌株之间基于k-mer的Jaccard相似性很高,StrainScan将大多数菌株分配到一个具有大量k-mers的大簇中(Supplementary Fig. S6),因此StrainScan需要更多的时间来区分它们。尽管如此,菌株扫描在结核分枝杆菌菌株鉴定方面仍然具有最好的召回率和准确性。所有应变识别实验均在配备2.4Ghz 14核Intel Xeon E5-2680v4 cpu和128 GB内存的HPCC CentOS 6.8节点上进行。我们为所有工具使用了8个线程。总之,strain scan能够在不牺牲分辨率的情况下实现更高的精度,即使真正的菌株具有高序列相似性的同行。
从模拟数据中检测共存菌株
研究表明,与人类相关的微生物群往往是同一物种密切相关菌株的复杂混合物[15]。为了定量比较Krakenuniq、StrainSeeker、StrainGE、StrainEst和StrainScan对同一物种多株菌株的识别性能,我们从6种细菌中随机选择2、3和5株菌株,生成模拟数据集。由于Sigma和Pathoscope2处理这些数据集的时间太长,因此没有纳入本实验。
为了研究菌株之间的相似性如何影响工具的性能,我们在选择多个菌株时使用了两种策略。在StrainScan的聚类步骤中,基于k-mer的Jaccard相似性大于或等于95%的菌株(对应于近似的ANI为99.89%)被分组到同一个聚类中。因此,在同一群集中共存的菌株比来自不同群集的菌株更难识别和区分。为了考虑不同的难度水平,我们的第一种策略从不同的集群中随机选择菌株,第二种策略从同一集群中选择不同的菌株。对于每种策略,我们随机选择2、3和5株菌株(3组),并使用不同的覆盖谱模拟短读:2株为100X和10X, 3株为100X、50X和10X, 5株为100X、70X、50X、20X和10X。其他读取的仿真参数与“单应变”实验相同。然后选择另一组菌株,重复实验10次。最终,对于每种细菌,我们使用第一种和第二种策略生成了30组包含不同数量菌株的数据,总共60组数据。因此,这六种细菌总共有360 (60 × 6)个模拟数据集。
多应变情况下的集群级性能评估如果一个样本包含n个来自stringe或StrainEst定义的同一集群的应变,则根据这些工具的设计,只会返回一个具有代表性的应变。使用这一具有代表性的菌株将导致多菌株实验的召回率非常小。为了避免这种情况,返回的代表性菌株将被计数n次,这通常使召回率为1.0,有利于这些工具。因为我们的簇比stringe和StrainEst定义的簇具有更大的粒度,所以从同一簇的菌株中模拟的样本都属于这种情况。
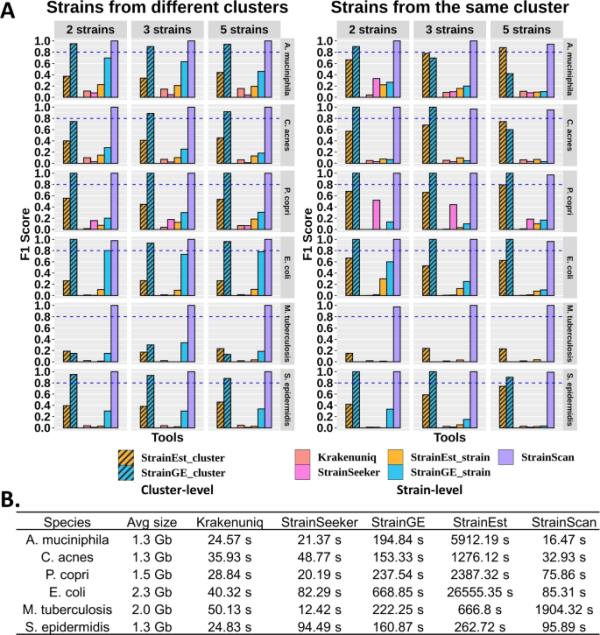
A 5种工具在“多应变”模拟数据集上的F1得分。标题中的“聚类”是指CST算法生成的聚类。共有60组模拟reads,分别包含2、3和5个菌株,每种细菌具有不同的相似性。注意,StrainSeeker无法识别结核分枝杆菌菌株,因此相关得分为0。B 5个被测工具的运行时间比较
不同工具的F1得分对比如图7A所示。TP、FN、FP、召回率和精密度记录在补充表S5和S6中。如图7A所示,StrainScan在所有测试数据集上都获得了近乎完美的F1分数。菌株在copri和大肠杆菌实验中具有与StrainScan相同的簇级F1分数。其簇级F1得分一般随着输入应变相似性的增加而降低,这可以从图7A左右两面板的对比中观察到。例如,其对嗜粘杆菌5个菌株数据集的聚类召回率从0.94下降到0.42(补充表S6)。与单菌株实验类似,stringe在集群水平上对M. tuberculosis的表现仍然很差。StrainEst的输出FPs较多,导致精度较低,F1得分较低。然而,由于我们评估簇级性能的方式,StrainEst对同一簇的菌株的簇级召回率似乎高于不同簇的菌株,从而获得更好的F1分数(图7A的右面板)。其余工具的F1得分一般较低。其中,Krakenuniq对不同菌群菌株的识别效果优于对同一菌群菌株的识别效果,这与Krakenuniq方法一致。StrainSeeker在所有被测数据集中的FPs都很高,对大多数被测细菌的召回率也很低,表明它不适合识别多个菌株。此外,通过分析相同分数的返回株数(StrainSeeker)和返回簇中的株数(StrainEst和StrainGE),我们还发现,菌株之间的高相似性进一步降低了StrainSeeker、StrainGE和StrainEst的分辨率(Supplementary Fig. S8)。总的来说,与其他测试工具相比,strain scan在保持高分辨率的同时,在所有数据集的应变水平上实现了F1分数的提高。
除了结核分枝杆菌外,StrainScan也比strain和StrainEst快(图7B)。如前一节所述,由于结核分枝杆菌菌株之间的高度相似性,StrainScan牺牲了在同一大簇中区分菌株的计算效率,因此需要更长的时间来处理结核分枝杆菌。
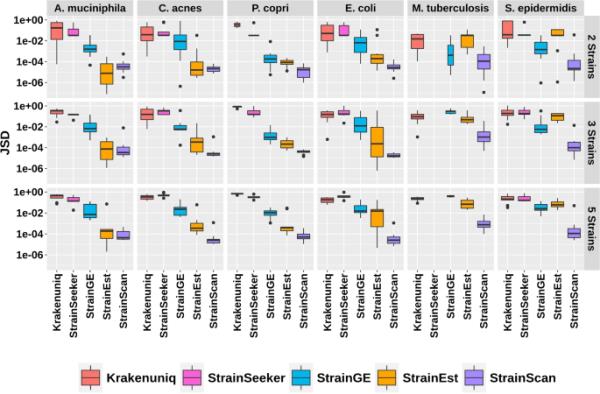
实际真实值与预测相对丰度之间5种工具的Jensen-Shannon散度(JSD)。因为stringe和StrainEst总是为来自同一簇的菌株返回一个代表性菌株,所以这里显示的结果只包括来自不同簇的菌株。包含同一簇和不同簇的菌株的完整结果见补充图S9
相对丰度计算为了衡量在合成数据集中预测应变曲线的准确性,我们计算了实际频率与推断频率之间的Jensen-Shannon散度(JSD)。如果预测的相对丰度和真实的相对丰度的维数可能不同,我们在维数较低的那一个上加零来计算JSD。在stringe和StrainEst的情况下,丰度是在集群水平上计算的。例如,如果样品中有两个菌株,并且这两个菌株具有相同的代表菌株,则我们使用代表菌株及其丰度两次来计算JSD。这将导致比将菌株的丰度之一设置为零更小的JSD。结果如图8所示。在所有情况下,strain scan重建的应变分布精度较高,中位数为Jensen-Shannon散度(JSD)。随着菌株数量的增加,strain和StrainEst在定量组成方面表现较差。对于含有高度相似或大量菌株的物种,如大肠杆菌、结核分枝杆菌和表皮葡萄球菌,StrainScan明显优于strain和StrainEst。对于StrainSeeker和Krakenuniq,大多数情况下JSD大于0.1,说明这些工具不能准确量化菌株的组成。随着应变数和相似性的增加,大多数测试工具的JSD值的中位数增加,而StrainScan的JSD在所有情况下波动不大。这些结果表明,即使对于含有高度相似菌株的样品,StrainScan也能比其他工具更好地定量复杂样品的组成。
为了评估不同工具在较低深度下识别多株菌株的能力,我们使用不同的覆盖谱对之前选择的2株菌株进行了额外的短读模拟。然后,我们使用这些数据集对所有工具进行基准测试。结果表明,StrainScan在从许多高度相似的菌株中识别低深度菌株方面具有明显的优势。例如,在识别10X和1X覆盖率的痤疮C.菌株时,StrainScan的F1得分为0.98,而排名第二和第三的Krakenuniq和StrainGE的F1得分仅为0.88和0.81(见补充表S7)。总体而言,StrainScan在识别低深度多重菌株方面表现出竞争力(补充表S7和S8)。关于这个实验的更多细节可以在补充章节2.1中找到。
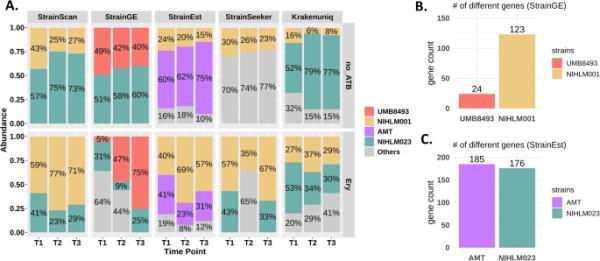
在90个模拟和30个真实的大肠杆菌数据集中,鉴定菌株与地面真实之间的捣碎距离。根据strain scan数据库中所有菌株与实际菌株之间的“1-Mash距离”将模拟数据集分为四组
如果实际应变不在参考数据库中怎么办se吗?
当数据库中不包含目标菌株时,我们期望菌株识别工具能够返回数据库中最匹配的菌株。为了评估不同工具在确定最佳匹配方面的性能,我们下载了NCBI于2022年发布的90个完整的大肠杆菌基因组(补充表S9)。由于所有工具的大肠杆菌参考数据库都是使用截至2021年的完整基因组构建的,因此所有这些基因组都不在我们构建的数据库中。然后,我们从90个大肠杆菌基因组中模拟了10倍覆盖率的读取,并将其作为所有工具的输入。对于每个数据集,图9中的“真值”是所有1433个大肠杆菌菌株中实际菌株与其最佳匹配之间的Mash距离(表2)。然后,我们在图9的前四个面板中记录了每个数据集的返回菌株与实际菌株之间的Mash距离。需要注意的是,这是一个单应变实验,因此Mash距离是用返回应变(包括代表应变)和实际应变计算的。90株中有54株的聚类在StrainScan的参考数据库中缺失。然而,StrainScan可以正确地识别出这54个菌株在数据库中最接近的匹配簇(Supplementary Table S9)。此外,与其他工具相比,strain scan识别的菌株与实际菌株的Mash距离更小。虽然stringe和StrainEst确定的最接近匹配在“1-MashD”范围为0.98到0.99时与“真相”的Mash距离相似,但当“1-MashD”增加时,它们的性能下降。例如,当实际应变与最佳匹配的“1- mashd”在(0.999,1)以内时,strain和strain est倾向于从较大的集群中返回一个具有代表性的应变(补充表S9)。结果,这些工具返回的代表性菌株具有比实际菌株更大的粉碎距离。相比之下,strain scan实现簇内应变识别,并返回更准确的最佳匹配。
为了进行更可靠的测试,我们下载了30个真实的大肠杆菌全基因组测序数据,并在NCBI中绘制了基因组草图。为避免数据相关偏倚,我们从3个不同的项目(PRJNA509690、PRJNA479542、PRJEB21464)中随机选取30个真实的测序数据集,覆盖范围为20X ~ 94X[46,47]。根据NCBI的说法,这些基因组草图的组装水平都是“脚手架”而不是“完成”。因此,所有这些基因组也不在使用完整基因组构建的大肠杆菌参考数据库中。我们进一步将这30个草案基因组的RefSeq加入与我们构建的数据库中的基因组进行比较,没有发现任何匹配。然后,我们将所有工具应用于这些真实数据集,并将已识别的菌株与草图基因组进行比较(图9的最后一个面板)。结果,StrainScan仍然比其他工具返回更准确的最佳匹配,证明了其在实际应用中的实用性。
考虑到数据库中缺乏参考序列的多个菌株可能在一个样本中共存,我们使用单菌株实验中的90个大肠杆菌基因组进行了额外的2菌株实验。结果表明,StrainScan在识别数据库中没有参考基因组的多个菌株时达到95%的F1得分,而第二好的工具stringe(集群级)仅达到78%的F1得分(见补充图S10)。此外,在所有测试数据集中,StrainScan没有产生任何假阳性鉴定,并且与其他工具相比,StrainScan鉴定的菌株与地面真实值的Mash距离更小(见补充表S10)。关于这个实验的更多细节可以在补充章节2.2中找到。
应变扫描的评估让我感到震惊tagenomic序列
虽然以前的实验主要使用模拟或真实的全基因组测序数据,但我们现在评估Strainscan是否在包含不同物种的宏基因组数据上保持相同的性能。为此,我们使用加尖的宏基因组数据进行了一项实验。具体来说,通过将模拟的copri和大肠杆菌数据集与真实数据混合,我们生成了130个加标的宏基因组数据,随后我们使用StrainScan对其进行分析。结果表明,StrainScan在这些加标宏基因组数据集上的结果与在模拟全基因组测序数据上的结果相同,证明了StrainScan在复杂样本上的鲁棒性(Supplementary Table S11)。关于这个实验的更多细节可以在补充章节2.3中找到。
e模拟数据上的StrainScan估值
在本实验中,我们对来自人类微生物组计划(Human Microbiome Project)的两个样本进行了StrainScan测试[49]。它们含有21种已知的具有均匀(SRR172902)或交错组成(SRR172903)的生物。在21种微生物中,3种细菌(大肠杆菌、痤疮C.和表皮葡萄球菌)是菌株水平分析的难点,我们已经建立了它们的参考索引结构。因此,我们使用StrainScan对这三种细菌进行了菌株水平分析。根据给定的数据描述,每个细菌在这两个数据集中只有1个菌株。虽然这是一种单应变检测,但这种测试具有挑战性,因为一些目标在样品中的丰度很低。为了比较,我们还使用Krakenuniq, StrainSeeker, StrainGE和StrainEst来鉴定这两个数据集中这些细菌的菌株。这5种工具的结果如表3所示。
对于所有被测试的物种,strain scan正确地识别出一个与真实菌株高度相似的优势菌株的存在(与真相的距离)。除了StrainScan, StrainSeeker和stringe还返回了与地面真实高度相似的应变。然而,StrainSeeker的输出通常包含多个相同分数的命中,这使得准确的评估变得困难。例如,它在两个测试数据集中返回119个大肠杆菌菌株,这使得用户很难知道这些样本中存在的实际菌株。在表3中,我们将与真实值的Mash distance最小的菌株作为strain seeker预测的优势菌株。StrainGE也返回与地面真实值有一个小的Mash距离的strain。在剩下的两个工具中,StrainEst无法识别低丰度菌株,并且需要很长时间才能运行,而Krakenuniq返回的结果与基本事实有很大不同。
为了评估每种工具在已知多菌株组成的真实样本上的性能,我们下载了一个模拟群落测序数据集(SRR13355226),该数据集包含大量来自宿主(即人类)以及四种不同的大肠杆菌菌株的reads。所有的读取都被用作所有工具的输入。strain scan是唯一在菌株水平上正确识别四种菌株的工具,没有假阳性鉴定(补充图S11)。菌株正确鉴定4种代表性菌株,无假阳性鉴定。然而,stringe鉴定的代表菌株与真实菌株之间存在136个不同的基因。基因预测采用Prokka[58],比较分析采用Roary[59]。尽管StrainEst也正确地识别了四种代表性菌株,但它返回了许多FPs。其余两种工具不能正确识别所有四种菌株,其中StrainSeeker的输出包含多个相同分数的匹配,而Krakenuniq报告了许多假阳性菌株。
strain scan从真实的测序数据中检测致病菌株
为了说明菌株扫描在病原体检测中的潜在应用,我们在两项研究中应用菌株扫描检测了大肠杆菌和结核分枝杆菌的致病菌株的存在(BioProject Accession: PRJEB1775和PRJEB2777)。第一项研究与2011年德国爆发的大肠杆菌疫情有关[50],该疫情是由肠聚集菌(EAEC)菌株引起的。这些数据集使用粪便样本的宏基因组测序进行测序。根据原文,致病菌株是大肠杆菌O104:H4。第二项研究调查了REMoxTB临床试验患者中结核分枝杆菌复发的频率,该试验评估了先前未经治疗的患者对结核分枝杆菌的治疗[51]。测序数据是从细菌分离物中获得的,每个数据集都有一个公开可用的组装基因组,代表样品中所含的菌株。从这两项研究中,我们分别选择了6个样本进行实验。为了比较,我们还应用其他工具对这些真实测序数据中的致病菌株进行检测。如表4所示,StrainScan能够在所有测试数据集中识别正确的菌株,而其他工具无法在某些数据集中检测正确的菌株。虽然strain est可以在大多数数据集中识别正确的菌株,但对于某些数据集,它只能返回正确菌株的代表性菌株。
菌株扫描准确地检测抗生素耐药的动态美国epidermidis菌株
在本实验中,我们测试了StrainScan在公共宏基因组数据集(PRJNA490375)中检测两种表皮葡萄球菌菌株动态的能力。根据最初的研究[52],这些数据集是由体外混合菌株培养产生的。两组分别以1:1的比例培养2株表皮葡萄球菌皮肤分离株。一组采用抗生素红霉素治疗(Ery),另一组不采用抗生素治疗(no_ATB)。其中NIHLM023对红霉素不耐药,而NIHLM001对红霉素有高度耐药。最后,通过宏基因组测序在三个不同的时间点从两组中获得6个数据集。虽然没有给出这两种菌株的相对丰度,但给出了各时间点的覆盖率,可以用来评价两种菌株的比例变化情况。根据原始研究报告的覆盖率,NIHLM023在no_ATB组中始终为优势菌株,而NIHLM001在Ery组中各时间点均为优势菌株。然后,我们对这六个数据集应用StrainScan等工具,结果如图10A所示。strain scan是唯一在每个时间点返回2个菌株正确的菌株比例,并且在所有样品中没有假阳性鉴定的工具。在测试工具中,StrainGE和StrainEst在某些样品中返回了正确的代表性菌株。因此,我们研究了这些代表性菌株与实际菌株之间的不同基因。基因预测由Prokka[58]完成,比较分析由Roary[59]完成。如图10B和C所示,这些菌株之间仍然存在许多不同的基因。在这些差异基因中,有些对菌株的功能非常重要。例如,NIHLM001具有ssaA_1基因,该基因已被证明与该菌株的许多特性相关,如耐药[60,61,62]。但代表性菌株UMB8493不具有该基因。理想情况下,对于更准确的应变水平分析,首选应变水平分辨率。
A 6个真实宏基因组样本中5种工具估计的表皮葡萄球菌菌株的丰度。No_ATB:未给药组,无耐药菌株NIHLM023为优势菌株。Ery:采用抗生素红霉素治疗组,耐药菌株NIHLM001为优势菌株。每种颜色代表一种菌株。B-C实际菌株与stringe和StrainEst鉴定的代表菌株之间的不同基因数
StrainScan识别低深度毒性梭状芽孢杆菌来自我的压力tagenomic数据
为了进一步测试StrainScan识别低深度致病菌株的能力,我们将StrainScan和其他工具应用于一个包含低深度毒力艰难梭菌菌株的元基因组数据集[53]。根据最初的研究,在该数据集中检测到两种低深度(1X)的强毒艰难梭菌菌株。在这两个菌株之间只报告了3个突变。另一项研究[63]也检测到艰难梭菌菌株之间存在相同的突变,这表明它们是多个高度相似的菌株存在的结果,而不是测序错误。由于艰难梭菌不是六个目标细菌之一,我们首先使用从NCBI RefSeq下载的102个完整艰难梭菌基因组构建了每个工具的参考数据库。strain scan、StrainGE、Krakenuniq、Pathoscope2和Sigma能够检测到约1倍深度的菌株(表5)。而Krakenuniq、Pathoscope2和Sigma检测到的菌株超过10株,说明存在大量假阳性。虽然菌株只输出一个菌株,但鉴定出的菌株不含致病性艰难梭菌应有的两个基因TcdA和TcdB。strain scan是唯一检测低深度毒株而无假阳性的工具。然而,由于strain scan和StrainGE的深度较低,与优势菌株的相似性极高,因此都错过了另一个菌株。
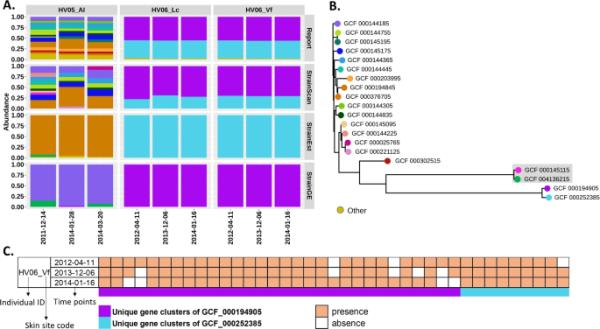
菌株扫描揭示了9个真实宏基因组样本中更大的多样性。这些样本取自两名健康人(HV05和HV06)在不同时间点的三个不同皮肤部位(AI、Lc和Vf)。在原文[15]中对站点代码进行了描述。B鉴定菌株的系统发育树。叶子使用与(A)相同的模式着色,距离为Mash距离[14]。该树通过iTOL可视化显示[65]。C“GCF_000194905”和“GCF_000252385”存在独特的基因簇。“GCF_000194905”和“GCF_000252385”高度相似,属于StrainGE和StrainEst的同一簇。存在是由原始研究[15]给出的,每一列表示一个独特的基因簇
StrainScan揭示了更大的多样性c·曼秀雷敦在人体皮肤上
既往研究[11,15]表明,痤疮C.是人类皮肤上最常见的细菌之一,通常具有复杂的多菌种群落。应用StrainEst重新分析研究中的人体皮肤数据集(SRP002480)[29]。然而,我们发现在一些样品中,StrainEst鉴定的菌株数量往往少于原始研究报告的菌株数量。在最初的研究中,作者使用内部管道来确定菌株并预测其在样品中的相对丰度,并且该内部管道的准确性先前已通过对人体皮肤微生物组数据的大量模拟来验证[11,15]。因此,我们从符合此情况的两个人中选择了9个样本,然后使用StrainEst和StrainScan对这些样本进行重新分析。然后,我们将他们的输出与原始研究中的报告进行了比较。考虑到stringe的竞争性能,我们也将其加入到比较中。为了保持一致性,我们选择了原始研究中使用的所有菌株,建立了StrainEst, StrainGE和StrainScan的新定制数据库,并使用这些新建立的数据库进行后续分析。结果如图11所示。StrainEst和StrainGE在所有测试数据集中只返回一到两个菌株,而根据原始研究报告了更多的菌株。与StrainEst和StrainGE相比,StrainScan显示出与报告结果更相似的相对丰度模式。此外,StrainScan还在“HV05_AI”样本中检测到一些在原始研究中未发现的菌株,这表明C. acnes具有更大的多样性。例如,通过StrainScan和strain /StrainEst分别鉴定出两个高度相似的菌株“GCF_000145115”和“GCF_004136215”。如图11B的灰框所示,它们在树中高度接近,“GCF_004136215”是strain和strain est中“GCF_000145115”的代表菌株。然而,它们有76种不同的基因。根据原研究[11]的SNP分析,在“2011-12-14”的“HV05_AI”样本中鉴定出22个“GCF_000145115”的独特SNP,而“GCF_004136215”的独特SNP未被检测到。因此,样品中更有可能存在“GCF_000145115”而不是“GCF_004136215”,这显示了StrainScan高分辨率的优势。
此外,StrainGE和StrainEst对同一样品“HV06”鉴定出不同的代表性菌株(图11A)。结果表明,不同的聚类策略和代表性菌株选择策略会影响聚类方法的识别结果。相比之下,StrainScan在这些样本中鉴定出两种高度相似的菌株“GCF_000194905”和“GCF_000252385”,这与报道的结果一致。不同的应变组成结果会影响下游分析。我们以“HV06_Vf”的分析结果为例。原始研究通过对“GCF_000194905”和“GCF_000252385”独特基因簇的reads比对,分析了“HV06_Vf”中这两株菌株的基因含量随时间的变化。如图11C所示,来自两个菌株的独特基因簇在不同时间点存在差异,这反映了该个体在菌株水平上的功能差异。这一结果表明,StrainScan可以区分高度相似的菌株,可以一起用于更全面的菌株水平功能分析。
两个例子展示了StrainScan在实际应用中的更多应用基因基因组测序数据
为了展示StrainScan在真实宏基因组测序数据中的广泛应用,我们应用StrainScan分析了横断面研究中的大肠杆菌菌株[48,54]和不同种群中的copri菌株[9]。分析结果表明,StrainScan可以从宏基因组样品中以更高的分辨率准确识别菌株,从而可以获得更全面的生物学见解。例如,StrainScan是唯一能够将来自三个国家的样品中的大肠杆菌菌株区分为三个不同组的工具(补充图S12)。同样,通过分析StrainScan鉴定的copri菌株,我们观察到杂食动物样本的菌株与素食动物样本的菌株在系统发育关系上明显分开,而素食动物样本的菌株介于两者之间(补充图S12)。关于这两个实验的更多细节可以在补充章节2.4和2.5中找到。
下载原文档:https://link.springer.com/content/pdf/10.1186/s40168-023-01615-w.pdf