


摘要
在这项工作中,评价和比较了混合模式色谱法和反相色谱法分离多肽和蛋白质消化。在三根具有反相/阴离子交换机制的混合模式色谱柱、两根反相十八烷基色谱柱和一根仅在限定pH范围内具有反相/阴离子交换特性的混合模式色谱柱上研究了流动相水相部分pH的影响以及有机改性剂对保留、分解和峰形的影响。选择极性、长度、氨基酸序列和电荷态不同的肽,即二肽、n -阻断二肽和寡肽,以恰当地描述不同条件下的色谱行为。这些测量显示了混合模式色谱柱在一次运行中分析不同电荷肽的潜力。通过对细胞色素C酶解片段的分析,验证了试验条件的适用性。分析比较了两种样品,即商业细胞色素C酶解标准品和胰蛋白酶自旋柱酶解细胞色素C。所得结果表明,由于细胞色素C酶解标准品中存在大量未知峰,可能是由胰凝乳和误裂解活性引起的,因此必须采用质谱检测。
图形抽象
介绍
混合模式色谱是一种很有前途的分析和分离多种化合物的工具[1,2]。其原因是分析物与固定相之间同时存在至少两种不同类型的相互作用,这会显著影响保留和分离[3]。因此,可以在一次色谱运行中分析各种不同的物理化学性质(如极性、电荷状态)的化合物[4]。这对于肽/蛋白质消化的分析是非常有利的,因为这些分析物通常是多电荷的,极性差异很大[5,6]。
混合模式色谱并不是一个全新的概念。例如,在离子交换色谱和亲和色谱中观察到“疏水相互作用”,在尺寸排除色谱中可能发生静电相互作用[7]。在混合模式色谱被认为是一种新的个体色谱方法之前,传统色谱模式中的二次相互作用(如来自解离的硅烷醇[8])通常被认为是不可取的——它们被认为是峰尾的主要原因,因此需要努力消除或至少最小化它们[9,10,11]。混合模式色谱与其他单一色谱模式的不同之处在于,它提供了两种或两种以上重要的不同类型的相互作用,因此所有的相互作用都有助于保留[3]。通常,混合模式色谱可以通过以下几种方法实现:(i)串联具有不同固定相/保留模式的两根色谱柱[12],(ii)在一根色谱柱中混合两种包装材料[13],(iii)配体链或载体中官能团的化学键合[14]。在一个固定相内对载体或配体进行不同类型官能团的共价修饰是目前获得混合模式固定相的主要方法。
在这项工作中,混合模式固定相结合了反相和阴离子交换保留机制。因此,分析物与C18配体之间的“疏水相互作用”,以及分析物与固定相带正电的部分之间的静电相互作用都有助于整体保留[15]。静电吸引或斥力主要由分析物官能团的pKa和流动相水相部分的pH决定[16]。因此,很明显,混合模式色谱提供了更多的可调流动相参数,这使得方法开发更加灵活,但也很复杂[17,18]。
具体来说,本研究使用了三种含有十八烷基配体和带正电改性剂的色谱柱,即:XSelect CSH C18柱的pyridyl基团,Atlantis PREMIER BEH C18 AX柱的季烷基胺,Luna Omega PS C18柱的带正电部分(详细信息未从制造商处获得)。色谱柱XSelect CSH C18作为一种反相色谱柱销售;然而,吡啶基的存在使固定相在流动相pH < 6时带正电荷[19]。另外两种混合模式色谱柱应该提供永久的正电荷,但我们之前的工作揭示了Luna Omega PS C18色谱柱与XSelect CSH C18色谱柱在与正电荷部分进行静电相互作用的pH范围方面的相似之处[5]。表1总结了本工作评估的固定相的示意图结构和基本性质。
混合模式固定相主要用于多肽和蛋白质等生物活性分子的分离[5,20,21,22,23]。对于更复杂的蛋白质分子,消化成更小的片段/肽是分析之前的必要步骤[1,24]。蛋白质的消化通常由胰蛋白酶完成,胰蛋白酶在蛋白质的c端精氨酸或赖氨酸残基处切割蛋白质(脯氨酸除外)[25]。胰蛋白酶消化可以通过几种方式进行,例如,在溶液中,通过胰蛋白酶自旋柱或在线(固定化胰蛋白酶反应器与液相色谱或毛细管电泳系统耦合)[26,27,28]。溶液消解需要缓冲液(Tris-HCl)、严格的pH值和温度控制(pH = 7.8,温度37.0°C)[29],并且非常耗时,通过自旋柱消解可能是一种合适的替代方法,它简单快捷[30]。
如上所述,测试的混合模式固定相结合了两种分离模式(反相和阴离子交换)的优点,并且能够根据分析物的极性和电荷同时分离复杂的混合物。出于这个原因,我们选择了一组不同性质(极性、电荷、长度、氨基酸序列)的肽来研究色谱参数对保留、分离和峰形的影响。采用单模C18柱进行比较。根据所得结果,选择了酶解细胞色素C的最佳分析条件。
结果与讨论
高效液相色谱法测量
首先,在高效液相色谱系统上对一组22种不同肽进行了初步测量。在xbridge C18(反相)、XSelect CSH C18 (pH < 6时混合模式)和Atlantis PREMIER BEH C18 AX(混合模式)三种不同色谱柱上研究了流动相水相pH对肽(二肽、n -阻断二肽和寡肽)的保留和分离的影响。本研究的目的是研究各种多肽在宽pH范围内的保留行为,以正确描述混合模式固定相,并比较混合模式色谱法和反相色谱法对多肽的保留率和选择性。
图1清楚地显示了使用梯度洗脱对各种多肽混合物分析的重要性,而二肽需要高含水量的流动相保留(95%的水相),阻断的二肽即使在由60-40%体积乙腈组成的流动相中也有足够的保留,这取决于所使用的色谱柱和流动相水部分的pH。阻断的二肽在n端含有苯甲酰保护基团,因此它们比未阻断的二肽更疏水。苯甲酰基团和C18配体之间的“疏水相互作用”(存在于每个测试的固定相中)与非阻断二肽相比具有更高的保留率。n阻断二肽羧基的pKa值在3.5-3.9范围内变化,即随着pH的增加,二肽的电荷越多,它们的极性越强,因此它们的log D值降低(支撑材料表S1)。这就是z - pH - pH - trp - oh在反相XBridge C18色谱柱上的保留率随pH值的增加而降低的原因(pH = 2.1和pH = 3.0的保留率相当)。对于柱XSelect CSH C18(图1B),情况非常相似,即使在pH < 6时固定相表面应带正电。事实上,在pH = 4.7时,CSH颗粒上的吡啶基仅部分带正电荷,因此更高的二肽极性优于静电吸引力(如[5]中已经显示的肽),并且保留率随着pH的增加而降低。
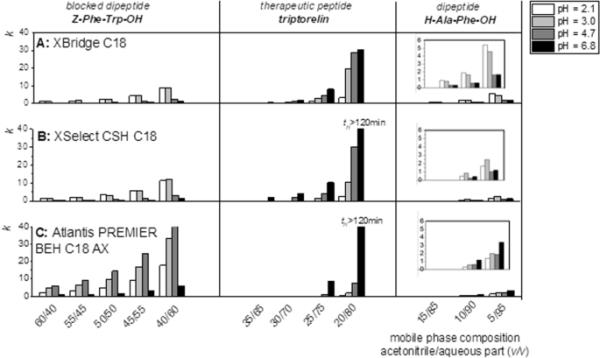
所选多肽代表(z - pH - trp - oh为阻断二肽,雷藤素为治疗性低肽,h - ala - phep - oh为二肽)的保留因子依赖于流动相乙腈含量(5-60体积百分比),流动相水相pH (pH = 2.1, pH = 3.0, pH = 4.7, pH = 6.8)和固定相(A: XBridge C18, B: XSelect CSH C18, C: Atlantis PREMIER BEH C18 AX)
混合模式色谱柱Atlantis PREMIER BEH C18 AX的情况并非如此(图1C),在pH = 4.7的流动相中观察到最高的保留率,其中带负电荷的n阻断二肽和带正电荷的固定相表面之间存在强静电相互作用。在pH = 6.8的流动相中,游离硅烷醇与游离硅烷醇发生静电斥力作用,滞留率下降。但与不带正电荷的固定相相比,保留率仍较高。混合模式色谱柱在流动相pH = 2.1时(无静电相互作用)n -阻断二肽的保留率高于其他色谱柱,这可能是由于孔径的差异(XBridge C18的孔径为130 × 10-10 m;选择CSH C18孔径130 × 10-10 m;Atlantis PREMIER BEH C18 AX孔径为95 × 10-10 m)。其他测试的n阻断二肽的保留趋势相同(图S1在支撑材料中)。非阻断二肽羧基的pKa值在3.5-3.8范围内,氨基的pKa值在8.0-8.5范围内(支撑材料表S1)。与n -阻断二肽相比,Log D值要低得多(为负,因为缺少苯甲酰),它们随pH的变化趋势正好相反,即随着pH的增加,Log D值也随之增加,流动相的水相pH = 4.7和pH = 6.8时,Log D值几乎相同(表S1)。这可能是由于电荷分布相似,在pH = 4.7的流动相中,所有的氨基都带正电,只有一部分羧基带负电。同样,在水相pH = 6.8的流动相中,所有羧基都带负电,只有一部分氨基带正电。这就是为什么在整个pH范围内,二肽在XBridge C18色谱柱上的保留率并没有下降,而只在pH = 4.7时下降(图1A)。在n阻断二肽的情况下,XSelect CSH C18柱上的保留行为与经典的反相柱非常相似(图1B)。在水相pH = 2.1的流动相中,固定相表面带正电的吡啶基与带正电的氨基之间的静电斥力进一步降低了本已很低的保留率。在pH = 4.7以上,固定相表面没有正电荷,因此只有带负电的羧基和带负电的自由硅烷醇之间的静电斥力起作用。这导致在所有测试的pH值中保留非常低。混合模式色谱柱Atlantis PREMIER BEH C18 AX随流动相水相pH值的增加而增加。在pH = 2.1的流动相中,由于带正电的氨基与带正电的固定相表面相互排斥,二肽未被保留。随着流动相水相pH值的增加,大部分羧基(能与带正电的固定相表面产生静电吸引)被解离,从而保持力增加。此外,游离硅烷醇基团可以与带电荷的氨基相互作用,从而增加了保留率。其他测试的非阻断二肽的保留趋势相似(支撑材料中的图S2)。
由于寡肽/治疗肽含有更多的功能基团,可以实现多次充电,因此对保留行为的描述更加复杂,并且对单个肽的描述也不同。当水相pH = 2.1时,所有三个测试柱在流动相均无寡肽保留(寡肽的最高极性,对数D值汇总于支撑材料中的表S1)。XBridge C18和XSelect CSH C18色谱柱提供了相同的趋势——雷普妥利林(图1)、leuprolide和戈舍林的保留率随着流动相pH的增加而增加。在支撑材料中,其他寡肽保留的个别趋势如图S3所示。混合模式色谱柱Atlantis PREMIER BEH C18 AX对所有测试的寡肽均显示出相同的趋势——随着流动相pH值的增加,保留率增加(图S3在支撑材料中)。
根据得到的结果,我们可以假设适当的条件分离所有测试肽的混合物,即二肽,阻断二肽和寡肽。结果表明,在pH = 2.1的流动相中,寡肽无滞留现象。此外,与pH = 6.8相比,pH = 2.1对n -阻断二肽的选择性非常低(图S4在支撑材料中)。因此,不使用水相pH = 2.1的流动相分析多肽混合物。对于非阻断二肽,使用较高pH的流动相水相(pH = 6.8)与混合模式色谱柱(如果排除pH = 2.1)相结合似乎是有利的。此外,这些条件也适用于分析阻断二肽。
图2显示了多肽混合物分离最佳结果的比较。XSelect CSH C18柱和Atlantis PREMIER BEH C18 AX柱在流动相水相pH = 6.8时,对大多数被测肽具有最高的选择性和合适的分辨率,与XBridge C18柱和其他被测流动相水相pH值相比。但是,可以清楚地看到,混合模式色谱柱Atlantis PREMIER BEH C18 AX提供了更高的保留时间,因此峰形较差(显然可以进一步优化)。
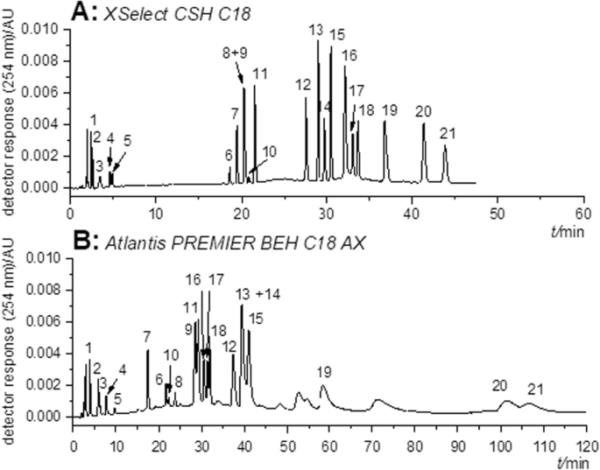
多肽混合物分离的比较。select CSH C18;B亚特兰蒂斯PREMIER BEH c18ax。梯度洗脱:乙腈/10 mM乙酸铵,pH = 6.8, 5/95 (v/v);t30min乙腈/ 10mm乙酸铵,pH = 6.8, 30/70 (v/v)。检测波长254nm。分析物:1:h - ala - tyroh;2: H-Tyr-Ala-OH;3: H-Val-Tyr-OH;4: H-Ala-Phe-OH;5: H-Phe-Ala-OH;6: [Met5]脑啡肽;7: [Lys8]后叶加压素;8: Z-Tyr-Ala-OH;9: [Leu5]脑啡肽;10:血管紧张素II;11: Z-Ala-Tyr-OH;12: Z-Ala-Phe-OH;13: Z-Ala-Trp-OH;14: Z-Phe-Ala-OH;15: Z-Trp-Ala-OH;16:戈舍瑞林;17: leuprolid;18: tritorelin;19: Z-Phe-Leu;20: Z-Trp-Phe-OH;21: Z-Phe-Trp-OH
进一步研究这两种色谱柱(XSelect CSH C18和Atlantis PREMIER BEH C18 AX)发现,对于细胞色素C消化片段的分析,Atlantis PREMIER BEH C18 AX柱具有更好的选择性(未观察到共洗脱)。(5)支持材料。
UHPLC测量
HPLC测量结果显示混合模式色谱分析多肽/细胞色素C消化的巨大潜力。因此,我们有兴趣进一步评估两种不同的混合模式色谱——luna Omega PS C18(类似于XSelect CSH C18柱,在pH > 6时失去正电荷)和Atlantis PREMIER BEH C18 AX(永久带正电荷)。为了确认混合模式色谱法的优势,我们还测试了反相色谱柱PREMIER BEH C18。由于UHPLC方法具有更高的效率,下面的实验是在UHPLC仪器上进行的,这也使我们能够分析一些其他的肽,这些肽是紫外检测不到的,因此需要不同的检测,在我们的情况下,质谱检测。为了证明混合模式固定相在多肽分离中的潜力,研究了流动相水相的pH值以及有机溶剂类型(甲醇与乙腈)对保留、分离和峰形的影响。测试了流动相水相部分的两种不同pH值:酸性pH值(0.1%甲酸溶液:26.5 mM, pH = 2.7,通常用于肽分析[33,32])和碱性pH值(26.5 mM甲酸铵,pH = 8.0,适合未来可能被胰蛋白酶在线消化蛋白质)。
图3证实了混合模式色谱法和反相色谱法之间的差异,以及不同混合模式色谱柱之间的差异(之前也在[5]中进行了描述)。对于所有测试的肽(图3A, B中所示的两个例子),与反相柱(甲醇和乙腈均适用)相比,流动相水溶液部分pH = 2.7的混合模式柱的保留率明显较低。其原因是主要带正电荷的肽与混合模式固定相表面之间的静电斥力。另一方面,在水相pH = 8.0时,观察到完全相反的趋势,即主要带负电荷的肽与带正电荷的混合模式固定相之间的静电吸引导致混合模式柱上的保留率高于反相柱;然而,Luna Omega PS C18与反相柱PREMIER BEH C18之间只有很小的差异(因为Luna Omega在pH > 6正电荷时损失)。此外,带负电荷的分析物与固定相表面解离的硅醇基团之间的静电斥力也有助于保留。总体的保留取决于自由剩余硅烷醇基团的数量,因此,保留是以下相互作用机制组合的结果-“疏水”相互作用,静电排斥和静电吸引。
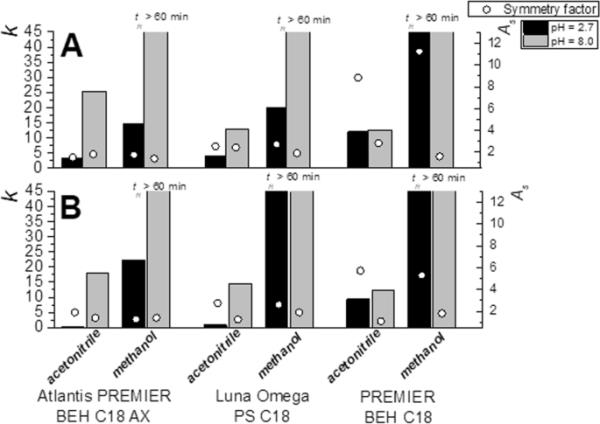
pH和有机溶剂对所有测试柱的保留率(k)和峰对称性(As)的影响。A: Val-Tyr-Val,流动相组成:有机溶剂/水相5/95 (v/v);B缓激肽,流动相组成:有机溶剂/水部分15/85 (v/v)
毫不奇怪,与乙腈相比,甲醇流动相的保留率明显更高。对比对称因子,甲醇和乙腈之间没有明显的差异(图3)。关于流动相水溶液部分pH对保留和峰对称的影响,我们观察到高pH (pH = 8.0)比低pH (pH = 2.7)提供更高的保留。这种影响在混合模式柱中非常显著,而在反相柱上的保留受流动相水相部分pH的影响不大,这表明固定相电荷的作用至关重要。此外,pH值越高,峰形越好(对称系数越低),其中对PREMIER BEH C18反相柱效果最为显著(图3)。
另一个目的是寻找在UHPLC系统中分离12种多肽混合物的合适条件。每个柱的大量梯度类型与每个水组分pH和每个有机改性剂的组合进行了测试。表2清楚地表明,在pH = 8.0的反相柱和Luna Omega PS C18柱上,实现所有感兴趣的肽的基线分离是最困难的。在这些条件下,不可能同时分辨出leuprolie - carbeoxytocin和angiotensin II-leucine - enkephalin这两对——梯度条件的变化导致其中一对或另一对的分辨能力丧失。但是,我们能够在混合模式色谱柱Atlantis PREMIER BEH C18 AX上基线分离所有肽,即使在使用甲醇流动相的5.5分钟内(获得的肽混合物分析最快见支撑材料中的图S6)。通常,在水相pH = 8.0的流动相中,Gly-Glu和Lys-Lys-Lys - lys的紫外检测信号很低(因为明显的峰劣化);因此,使用质谱检测是非常有利的。
选择细胞色素C作为模型蛋白,是因为有其酶解物的标准物,可以将酶解物的标准物与自旋柱酶解的细胞色素C进行比较。标准中声明的细胞色素C消化产物清单及相应的m/z值见支撑材料表S3。从高效液相色谱测量中发现,适合多肽分离的条件可能不适合同时分离细胞色素C消化酶。因此,细胞色素C消化物的分析(来自消化的标准物,通过自旋柱消化获得)在先前测试的UHPLC条件下进行,即三柱,乙腈与甲醇,水溶液部分pH = 2.7 vs. pH = 8.0,以及大量梯度(与用于分析12个肽的梯度不同)。
表2总结了获得的结果,即是否有可能基线分离所有细胞色素C消化和最短的分析时间。使用混合模式色谱柱Atlantis PREMIER BEH C18 AX(流动相为乙腈和pH = 2.7的水相)获得细胞色素C消化片段的最快分析,其中标准证书中声明的所有13个片段在11分钟内基线分离(图4A)。图4B为不同流动相(但同一固定相)的对比,即以甲醇为流动相,水溶液pH = 8.0。这些条件可能对在线蛋白质消化非常有利。对比图4A、B可知,在两种条件下,我们都可以实现13个片段的基线分离,只是洗脱顺序不同(流动相pH值不同)。
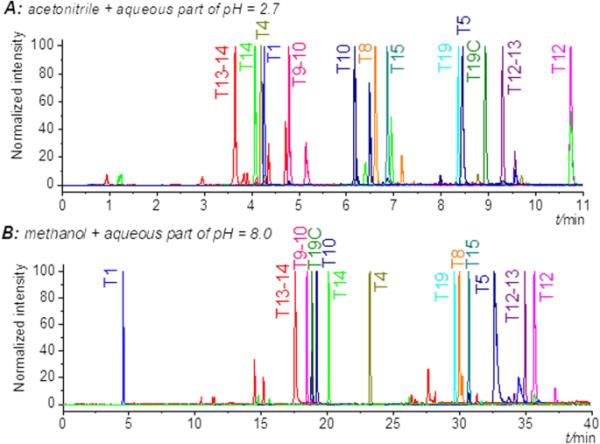
亚特兰蒂斯PREMIER BEH c18ax色谱柱分离13个细胞色素C酶切片段。A流动相A - 26.5 mM甲酸乙腈,B-26.5 mM甲酸水溶液,pH = 2.7,梯度洗脱:0 min-0% A;8 min-25% A;10.5 min-30% A;B流动相A - 26.5 mM甲酸铵甲醇,流动相B - 26.5 mM甲酸铵水,pH = 8.0,梯度洗脱:0 min-0% A;40分钟- 75% A
片段T19C源于胰凝乳促胰蛋白酶活性(表S3在支持物质中,氨基酸序列不以赖氨酸或精氨酸结束),可能在胰蛋白酶消化过程中由于不纯净的胰蛋白酶或胰蛋白酶自溶而产生[33]。通过自旋柱消化的细胞色素C样品中未发现该片段,这符合制造商的说法,即胰蛋白酶自旋柱纯化程度高,固定化可防止胰蛋白酶自溶[34]。T9-10, T12-13和T13-14片段是错切活性的结果,它们的存在在细胞色素C酶切的标准品中被声明,并且在通过自旋柱酶切的样品中也被观察到。
缺失的胰凝块并不是细胞色素C酶解标准品与自旋柱酶解细胞色素C的唯一区别。对这两种样品(消化标准品和通过自旋柱消化的样品)的分析比较显示,细胞色素C消化标准品的峰数增加,这表明了杂质的数量(图5)。消化蛋白质样品中的杂质通常是胰凝血或错切活性的结果,这在使用溶液内消化时很常见,特别是在消化时间延长时[35]。另一方面,通过自旋柱的消化可以达到较低的胰凝乳和误裂解活性。获得的数据表明,在没有质谱检测的情况下,蛋白质消化样品的分析存在问题。除了糜溶峰和杂质数量外,自旋消化法和消化标准品之间没有显著差异,即12个片段的保留率相当。
.jpg)
A细胞色素C酶解标准品与胰蛋白酶自旋柱酶解B细胞色素C紫外检测(214 nm)信号的比较色谱柱:Luna Omega PS C18,流动相A - 0.1%甲酸乙腈,流动相B-0.1%甲酸水溶液,梯度洗脱:0 min-0% A;8 min-25% A;10.5 -30%;12 min-40% A.支撑材料中的表S3包含了识别单个碎片的信息
目录
摘要 介绍 结果与讨论 结论 实验 数据可用性 参考文献 致谢 作者信息 补充信息 搜索 导航 #####结论
我们研究的目的是显示混合模式色谱分析多肽和蛋白质消化的巨大潜力。几种色谱柱,包括混合模式和反相色谱柱,在HPLC和UHPLC系统中使用。考察了流动相水相pH和有机改性剂种类对保留率、选择性、分辨率和峰形的影响。
对易于定义电荷态的二肽的分析有助于描述混合模式保留行为。结果表明,混合模式色谱柱Atlantis PREMIER BEH C18 AX在pH = 4.7的流动相水溶液中具有最高的负电荷二肽保留率。在较高的pH值下,带负电的残余硅烷醇的静电斥力占优势。证实了XSelect CSH C18色谱柱(以反相销售)仅在pH = 2.1和pH = 3.0的水相流动相中具有显著的混合模式特征。采用高效液相色谱法,在XSelect CSH C18柱上实现了21种多肽的基线分离。在峰形方面,发现酸性pH(2.7)条件下的PREMIER BEH C18柱最不合适(与乙腈流动相的对称系数大于8,与甲醇流动相的对称系数更高)。
UHPLC对细胞色素C酶切物的分析指出了质谱检测的必要性,因为细胞色素C酶切物标准品中存在大量的错切/糜溶片段。实验结果表明,Atlantis PREMIER BEH c18ax色谱柱的通用性最强。流动相的酸性部分(pH = 2.7)和碱性部分(pH = 8.0)的乙腈和甲醇均适用于细胞色素C消化酶的基线分离。混合模式柱Luna Omega PS C18除流动相为甲醇和甲酸铵缓冲液(pH = 8.0)外,结果相似。
实验
化学品和材料
乙腈(LC - ms级)和甲醇(LC - ms级)由VWR International (Radnor, USA)提供,LC用水购自Honeywell (Charlotte, USA),用于UHPLC测量。乙腈(Chromasolv®梯度级,高效液相色谱,≥99.9%)、甲醇(Chromasolv®梯度级,高效液相色谱,≥99.9%)、乙酸铵(纯度≥98%)、甲酸铵(纯度≥97%)、甲酸(纯度≥95%)、乙酸(纯度≥99%)、氢氧化铵溶液(28.0-30.0% NH3)和三氟乙酸(纯度99%)购自Sigma-Aldrich (St. Louis, USA)。去离子水用来自Watrex (Prague, Czech Republic)的Rowapur和Ultrapur系统纯化。除z - ph - leu和H-Val-Tyr由Sigma-Aldrich (St. Louis, USA)提供外,所有二肽均购自Bachem (Bubendorf, Switzerland)。醋酸戈舍林盐、醋酸leuprolide盐、醋酸缓激肽盐和卡霉素购自Bachem(瑞士布本多夫)。Lys-Lys-Lys、Val-Tyr-Val、血管紧张素I、血管紧张素II、[Lys8]-加压素、[Met5]-脑磷素醋酸盐水合物和[Leu5]-脑磷素醋酸盐水合物由Sigma-Aldrich (St. Louis, USA)提供。细胞色素C消解标准品由Waters (Milford, USA)提供。牛心脏细胞色素C(纯度> 95%)、胰蛋白酶自旋柱和4型蛋白提取试剂购自Sigma-Aldrich (St. Louis, USA)。所有测试分析物的清单、结构、log D和pKa值见支撑材料表S1。使用Marvin软件(ChemAxon公司产品)计算流动相水相对应pH值的log D值和多肽的pKa值。
仪器和色谱条件-高效液相色谱测量
所有HPLC测量使用Waters Alliance系统(Waters, Milford, USA),该系统由2690 D分离模块,2487双λ吸光度检测器,717 Plus自动进样器和Waters Alliance系列柱加热器组成。采用Empower 2软件进行系统控制和数据采集。使用的列有:XSelect CSH C18、XBridge C18和Atlantis PREMIER BEH C18 AX。所有测试柱,粒径为5 μm, 150 × 4.6 mm,来自Waters (Milford, USA)。
大多数分析物的原液是通过将样品溶解在浓度为1mg cm−3的甲醇中制备的。更多极性肽(H-Val-Tyr-OH, H-Ala-Tyr-OH, H-Tyr-Ala-OH)和血管紧张素II在浓度为1 mg cm−3的甲醇和水的混合物中(50/50 (v/v))溶解。由于极低的紫外响应,几种肽(z - ala - pheoh, z - ala - tyroh, z - phee - ala - oh, z - phee - leu和Z-Tyr-Ala-OH)需要在5 mg cm−3的较高浓度下溶解。第一个系统峰值被用作死时间标记。所有测量均为三份。
流动相为乙腈与水相,体积比为5/95 ~ 60/40 (v/v),采用5个体积百分比步长。对于多肽和细胞色素C消化物的混合物的分析,采用梯度洗脱。采用流动相的水相部分:365 mM甲酸,pH = 2.1;10 mM甲酸铵缓冲液,pH = 3.0;10 mM醋酸铵缓冲液,pH = 4.7, pH = 6.8。使用PeakMaster软件计算缓冲液成分浓度和相应的pH值[36]。设置基本色谱条件:流动相流速1 cm3 min - 1,进样量5 mm3,柱温25.0℃,样温20.0℃,检测波长254 nm和280 nm。细胞色素C酶切分析,进样量为15 mm3,检测波长为214 nm。
仪器和色谱条件- uhplc测量
使用Waters Acquity UPLC h级系统(Waters, Milford, USA)进行UHPLC测量。该系统配备了四元溶剂管理器、自动进样器、柱恒温器、光电二极管阵列检测器和QDa质量检测器。Empower 3软件用于系统控制、数据采集和结果处理。测试柱如下:Atlantis PREMIER BEH C18 AX;PREMIER BEH C18 (Waters, Milford, USA),两柱尺寸均为100 × 2.1 mm;粒径1.7 μm;柱Luna Omega PS C18 (Phenomenex, Torrance, USA),尺寸100 × 2.1 mm;粒径1.6 μm。
将样品溶解在浓度为1mg cm−3的去离子水中制备肽的原液。第一个系统峰值被用作死时间标记。所有测量均为三份。
流动相由乙腈或甲醇与水相组成,体积比为0/100 ~ 60/40 (v/v),分5步进行。对于多肽和细胞色素C消化物的混合物,采用梯度洗脱。流动相(B)的水相为0.1%甲酸,pH = 2.7(相当于26.5 mM的甲酸溶液)。同样,在pH = 8.0的水溶液中加入氢氧化铵,制得26.5 mM甲酸铵,使pH = 8.0。在这两种情况下,加入流动相B的甲酸/甲酸铵+氢氧化铵的量与加入流动相A的量相同(在梯度过程中保持离子强度恒定)。流动相A含有有机溶剂——纯甲醇(含甲酸或甲酸铵+氢氧化铵)、纯乙腈(含甲酸)或乙腈/水的混合物,80/20 (v/v)(甲酸铵+氢氧化铵),因为甲酸铵在纯乙腈中的溶解度很低。设置基本色谱条件:流动相流速0.3 cm3 min - 1,进样量1 mm3,柱温37.0℃,样品温度10.0℃,检测波长214 nm和220 nm + QDa检测(正模式,锥电压15 V,探头温度600℃)。
细胞色素C胰蛋白酶消化
对两种细胞色素C酶切样品进行了分析:(1)酶切细胞色素C标准品,(2)自旋柱酶切细胞色素C。将消化后的细胞色素C标准品溶于0.055%三氟乙酸200mm3中。自旋消解完全按照胰蛋白酶自旋柱(Sigma-Aldrich)附带的手册进行。自旋柱酶解包括以下步骤:蛋白质变性(使用尿素,硫脲和清洁剂的混合物,4型蛋白质提取试剂),自旋柱洗涤和平衡(使用100 mM碳酸氢铵反应缓冲液是自旋柱包装的一部分),酶解本身(加入100µg蛋白质,酶解15分钟)。消解产物用去离子水从自旋柱中洗涤,样品准备用于LC分析。
补充信息
以下是电子补充材料的链接。